

Alveolar inflammation in cystic fibrosis
- PMID: 20347403
- PMCID: PMC2883667
- DOI: 10.1016/j.jcf.2010.03.001
Alveolar inflammation in cystic fibrosis
Abstract
Background: In infected lungs of the cystic fibrosis (CF) patients, opportunistic pathogens and mutated cystic fibrosis transmembrane conductance regulator protein (CFTR) contribute to chronic airway inflammation that is characterized by neutrophil/macrophage infiltration, cytokine release and ceramide accumulation. We sought to investigate CF lung inflammation in the alveoli.
Methods: Lung tissue from 14 CF patients and four healthy individuals was analyzed for numbers of effector cells, elastin and collagen concentrations, inflammatory markers and density of Pseudomonas aeruginosa. Additionally, desmosine and isodesmosine concentrations were determined in 52 urine specimens from CF patients to estimate the burden of elastase activities in respiratory secretions.
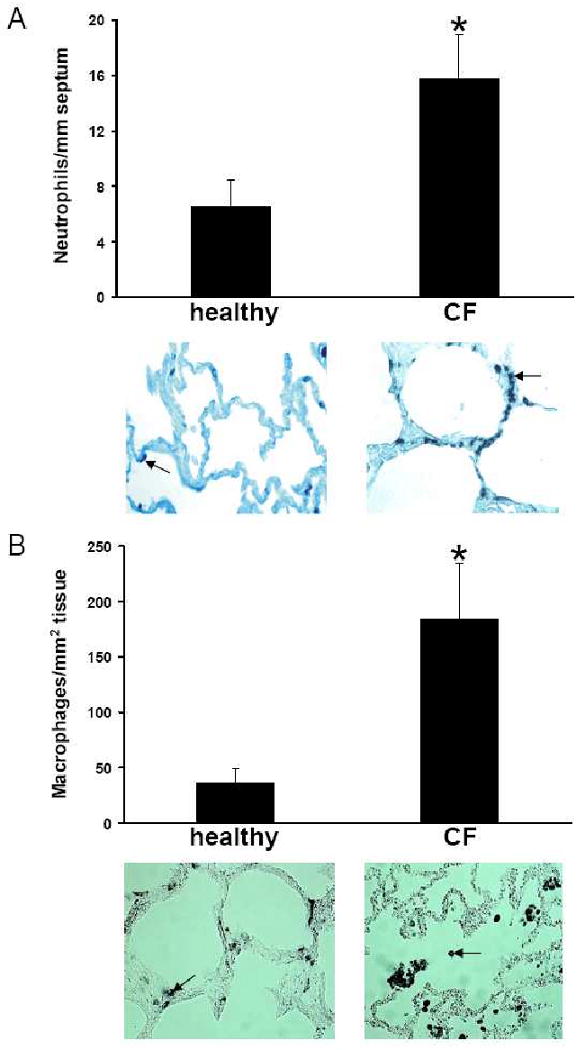
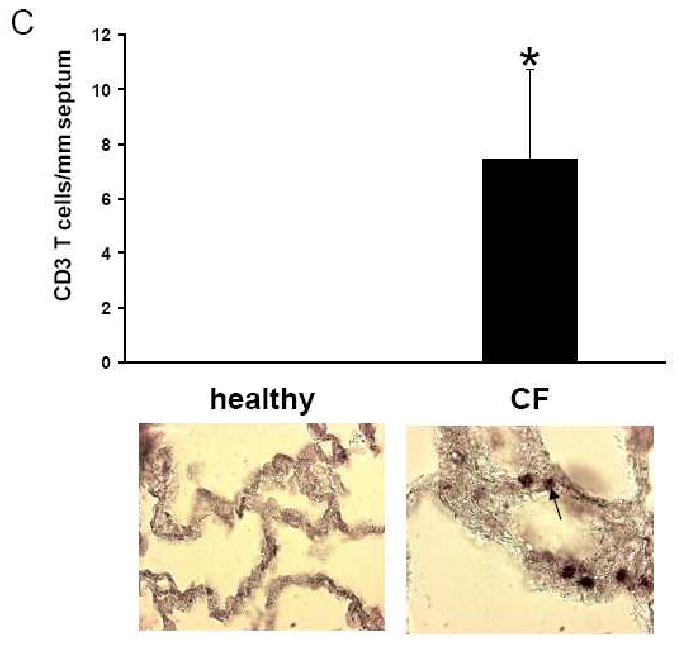
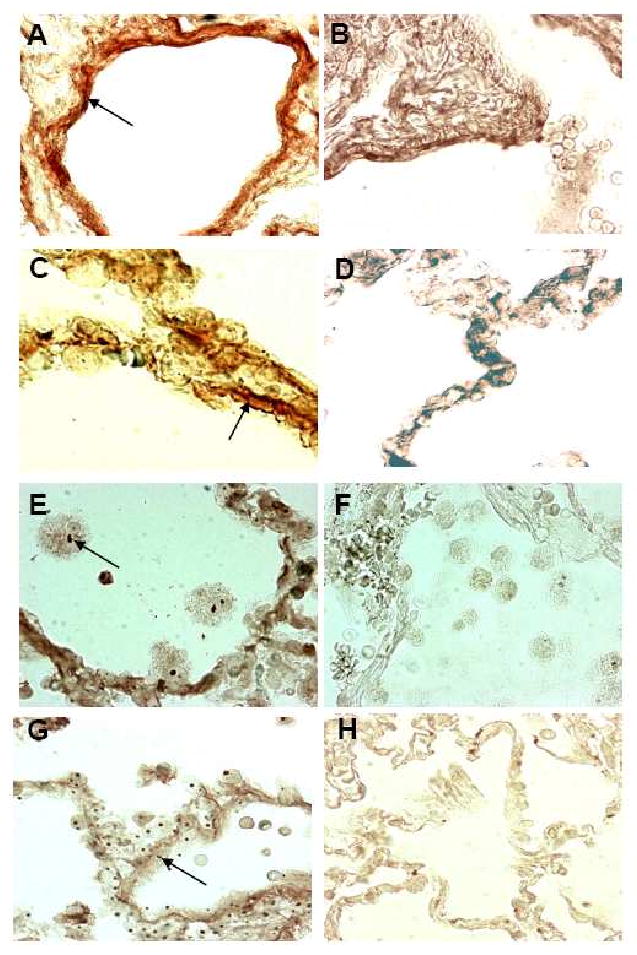
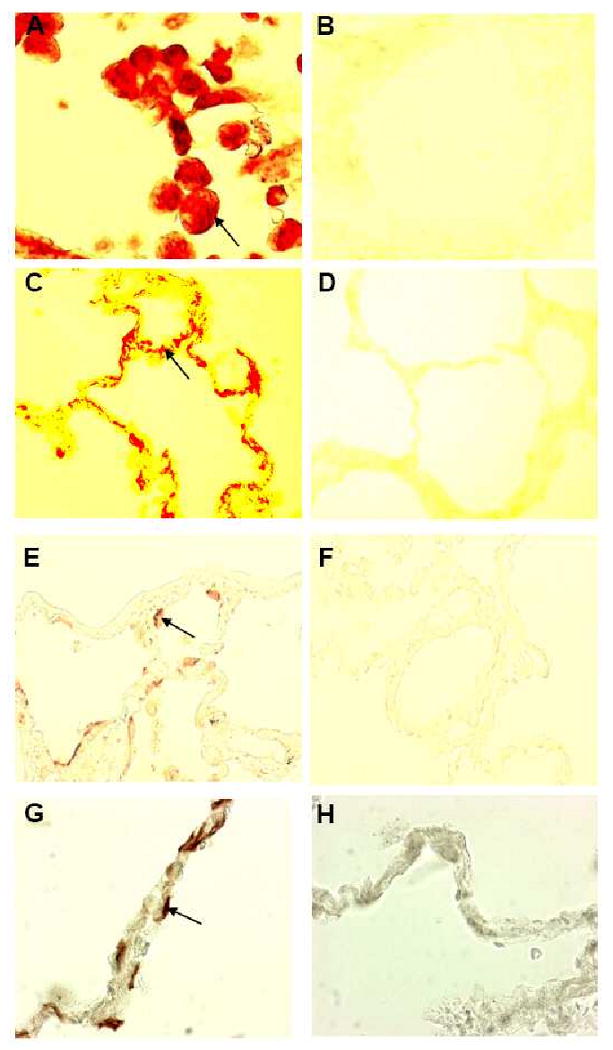
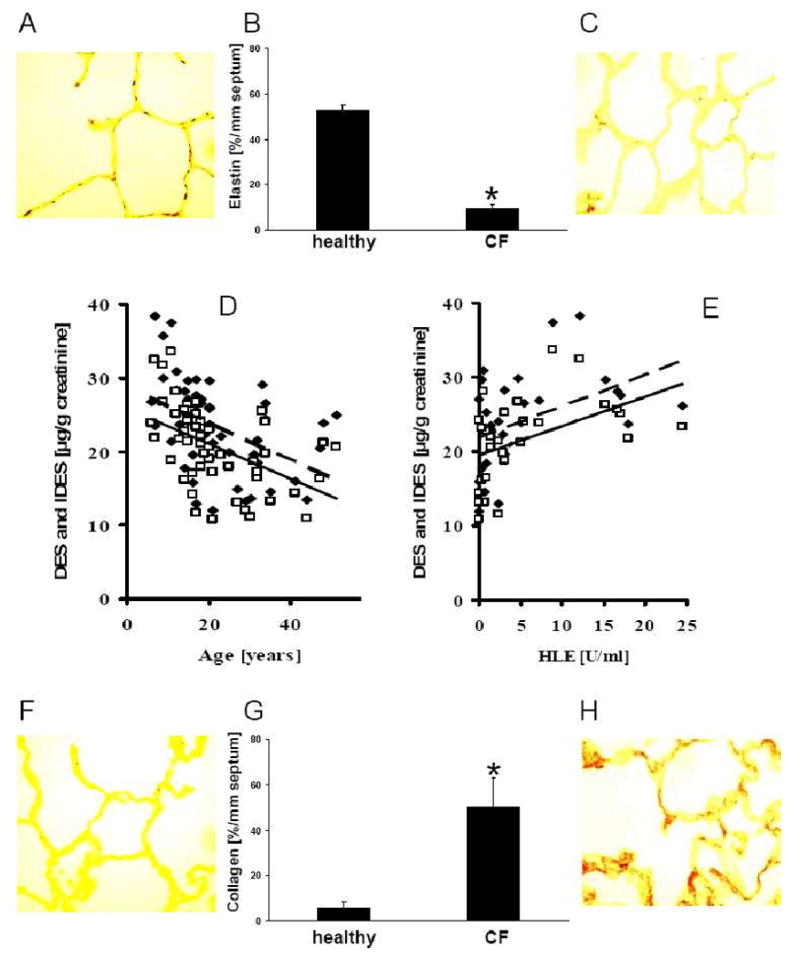
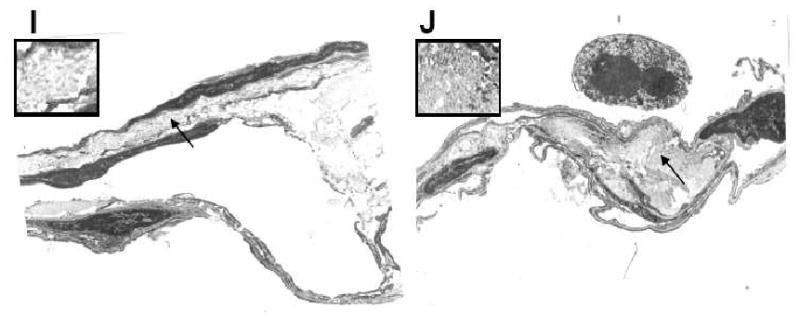
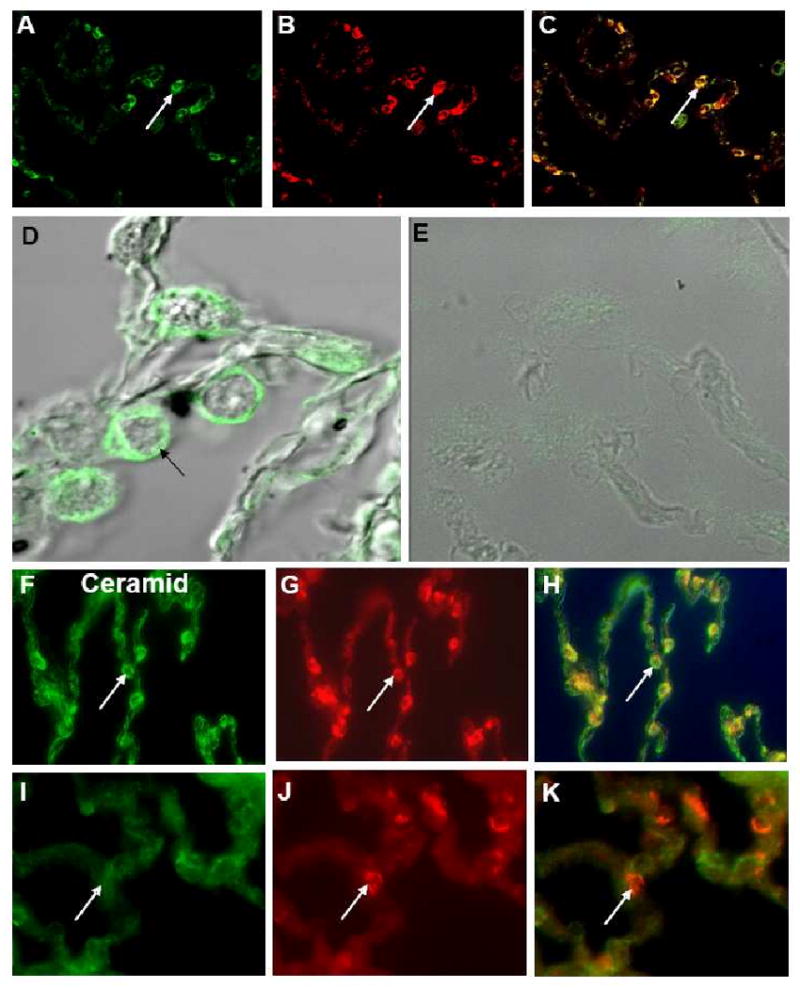
Results: Elastin concentration was significantly decreased and collagen significantly increased in CF alveolar tissues as compared to age-matched, healthy individuals. Elastin split products were significantly increased in urine samples from patients with CF and correlated inversely with age, indicating local tissue remodelling due to elastin degradation by unopposed proteolytic enzymes. Alveolar inflammation was also characterized by a significant cell infiltration of neutrophils, macrophages and T cells, extensive nuclear factor-kappaB and insulin-like growth factor-1 activation in various cell types and increased intercellular adhesion molecule-1 expression, and increased numbers of myofibroblasts. Additionally, ceramide accumulated in type II alveolar epithelial cells, lacking CFTR. P. aeruginosa organisms were rarely present in inflamed alveoli.
Conclusions: Chronic inflammation and remodeling is present in alveolar tissues of the CF lung and needs to be addressed by anti-inflammatory therapies.
Figures
References
-
- Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N, Zsiga M, Buchwald M, Riordan JR, Tsui LC, Collins F. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–65. - PubMed
-
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, Drumm ML, Iannuzzu MC, Collins FS, Tsui LC. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–73. - PubMed
-
- Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245:1073–80. - PubMed
-
- Boucher RC. New concepts of the pathogenesis of cystic fibrosis lung disease. Eur Respir J. 2004;23:146–58. - PubMed
-
- Worlitzsch D, Tarran R, Ulrich M, Schwab U, Cekici A, Meyer KC, Birrer P, Bellon G, Berger J, Weiß T, Botzenhart K, Yankaskas JR, Randell S, Boucher RC, Döring G. Reduced oxygen concentrations in airway mucus contribute to the early and late pathogenesis of Pseudomonas aeruginosa cystic fibrosis airways infection. J Clin Invest. 2002;109:295–09. - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
