

Inhibition by cigarette smoke of nuclear factor-κB-dependent response to bacteria in the airway
- PMID: 20348206
- PMCID: PMC3049229
- DOI: 10.1165/rcmb.2009-0454OC
Inhibition by cigarette smoke of nuclear factor-κB-dependent response to bacteria in the airway
Abstract
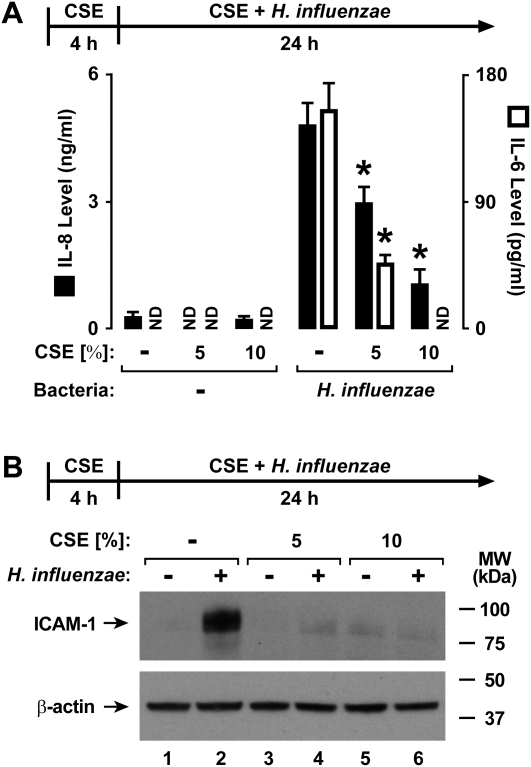
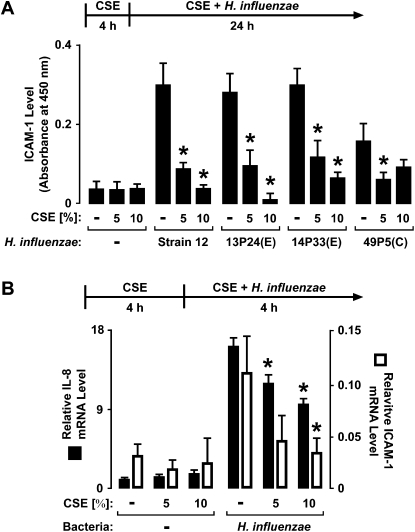
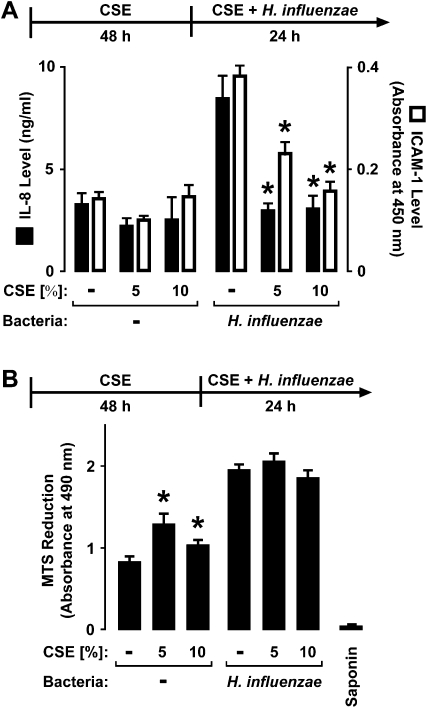
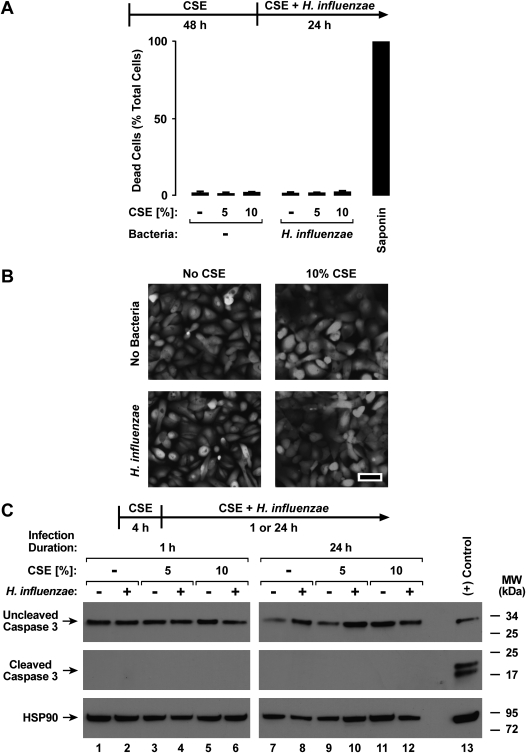
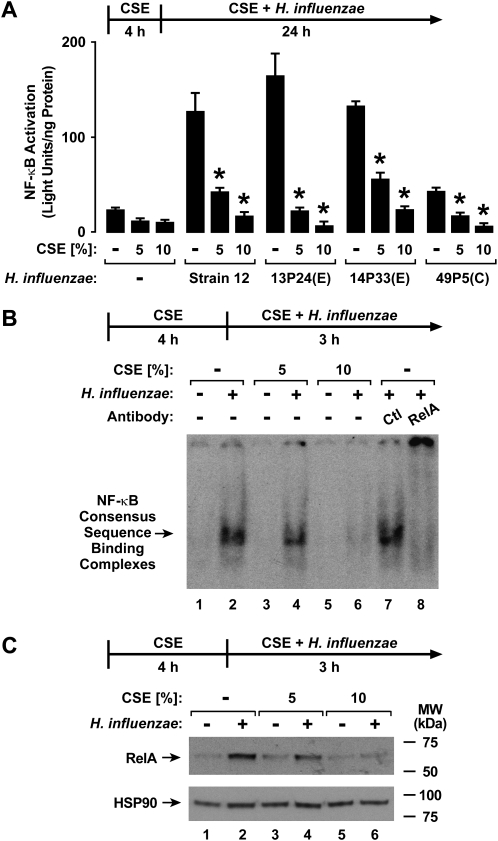
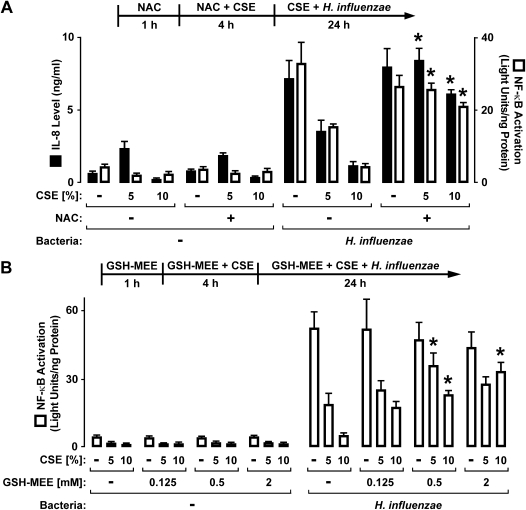
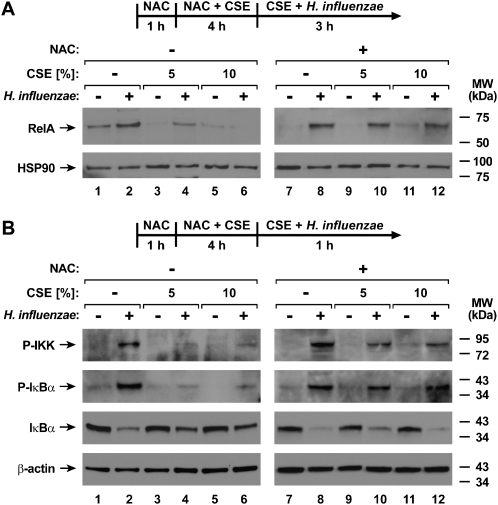
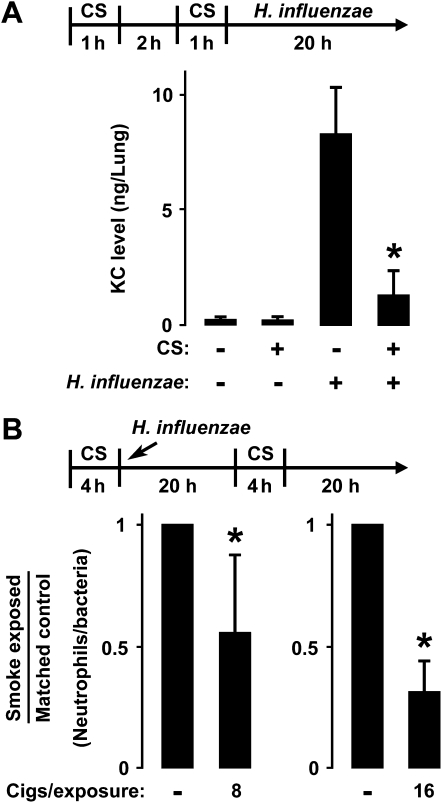
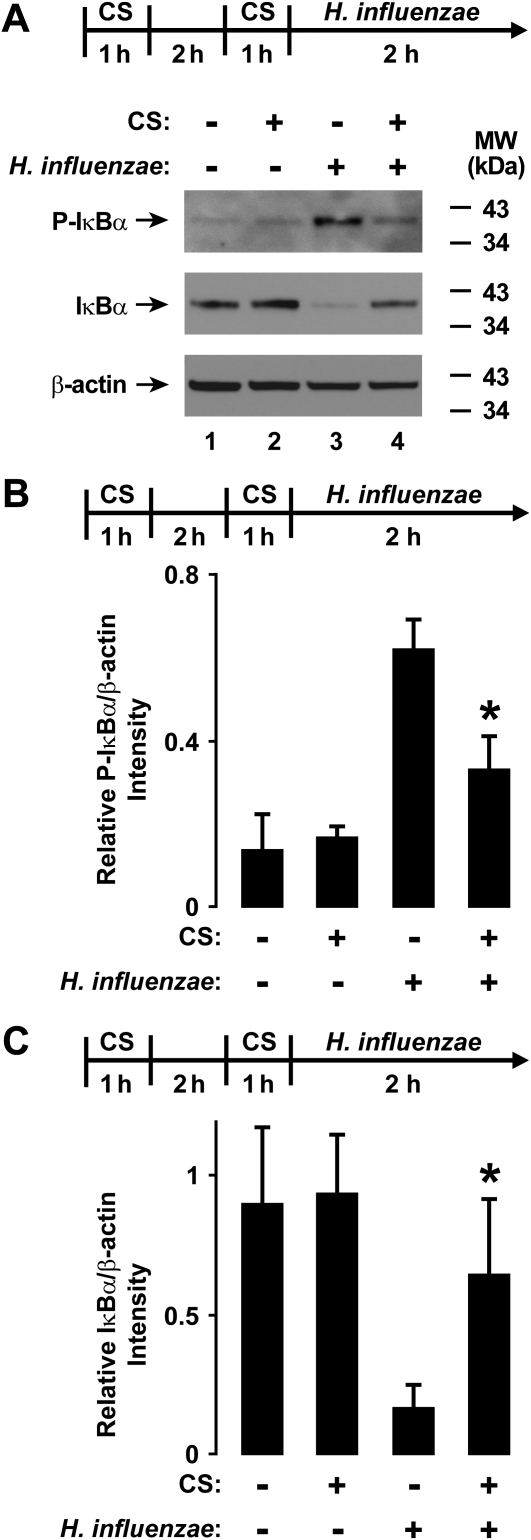
Although individuals exposed to cigarette smoke are more susceptible to respiratory infection, the effects of cigarette smoke on pulmonary defense are incompletely understood. Based on the observation that interactions between bacteria and host cells result in the expression of critical defense genes regulated by NF-κB, we hypothesized that cigarette smoke alters NF-κB function. In this study, primary human tracheobronchial epithelial cells were treated with cigarette smoke extract (CSE) and exposed to Haemophilus influenzae, and the effects of CSE on bacteria-induced signaling and gene expression were assessed. CSE inhibited high concentrations of induced NF-κB activation and the consequent expression of defense genes that occurred in airway epithelial cells in response to H. influenzae. This decreased activation of NF-κB was not attributable to cell loss or cytotoxicity. Glutathione augmentation of epithelial cells decreased the effects of CSE on NF-κB-dependent responses, as well as the effects on the inhibitor of κB and the inhibitor of κB kinase, which are upstream NF-κB regulators, suggesting the involvement of reactive oxygen species. The relevance of these findings for lung infection was confirmed using a mouse model of H. influenzae airway infection, in which decreased NF-κB pathway activation, keratinocyte chemoattractant (KC) chemokine expression, and neutrophil recruitment occurred in animals exposed to cigarette smoke. The results indicate that although cigarette smoke can cause inflammation in the lung, exposure to smoke inhibits the robust pulmonary defense response to H. influenzae, thereby providing one explanation for the increased susceptibility to respiratory bacterial infection in individuals exposed to cigarette smoke.
Figures
Similar articles
-
Inhibition of IFN-gamma-dependent antiviral airway epithelial defense by cigarette smoke.Respir Res. 2010 May 26;11(1):64. doi: 10.1186/1465-9921-11-64. Respir Res. 2010. PMID: 20504369 Free PMC article.
-
Cigarette smoke primes the pulmonary environment to IL-1α/CXCR-2-dependent nontypeable Haemophilus influenzae-exacerbated neutrophilia in mice.J Immunol. 2014 Sep 15;193(6):3134-45. doi: 10.4049/jimmunol.1302412. Epub 2014 Aug 4. J Immunol. 2014. PMID: 25092891
-
Cigarette smoke inhibits airway epithelial cell innate immune responses to bacteria.Infect Immun. 2010 May;78(5):2146-52. doi: 10.1128/IAI.01410-09. Epub 2010 Mar 1. Infect Immun. 2010. PMID: 20194598 Free PMC article.
-
Synergistic and feedback signaling mechanisms in the regulation of inflammation in respiratory infections.Cell Mol Immunol. 2012 Mar;9(2):131-5. doi: 10.1038/cmi.2011.65. Epub 2012 Feb 6. Cell Mol Immunol. 2012. PMID: 22307042 Free PMC article. Review.
-
Epithelial cells, the "switchboard" of respiratory immune defense responses: effects of air pollutants.Swiss Med Wkly. 2012 Jul 31;142:w13653. doi: 10.4414/smw.2012.13653. eCollection 2012. Swiss Med Wkly. 2012. PMID: 22851042 Free PMC article. Review.
Cited by
-
Inflammasome Activity in Non-Microbial Lung Inflammation.J Environ Immunol Toxicol. 2014 Sep 20;1(3):108-117. J Environ Immunol Toxicol. 2014. PMID: 25642415 Free PMC article.
-
In vivo Cigarette Smoke Exposure Decreases CCL20, SLPI, and BD-1 Secretion by Human Primary Nasal Epithelial Cells.Front Psychiatry. 2016 Jan 13;6:185. doi: 10.3389/fpsyt.2015.00185. eCollection 2015. Front Psychiatry. 2016. PMID: 26793127 Free PMC article.
-
How Do Innate Immune Cells Contribute to Airway Remodeling in COPD Progression?Int J Chron Obstruct Pulmon Dis. 2020 Jan 10;15:107-116. doi: 10.2147/COPD.S235054. eCollection 2020. Int J Chron Obstruct Pulmon Dis. 2020. PMID: 32021149 Free PMC article. Review.
-
Inflammasome involvement in CS-induced damage in HaCaT keratinocytes.In Vitro Cell Dev Biol Anim. 2022 Apr;58(4):335-348. doi: 10.1007/s11626-022-00658-x. Epub 2022 Apr 15. In Vitro Cell Dev Biol Anim. 2022. PMID: 35428946 Free PMC article.
-
Inflammatory and cytotoxic effects of acrolein, nicotine, acetylaldehyde and cigarette smoke extract on human nasal epithelial cells.BMC Pulm Med. 2014 Mar 1;14:32. doi: 10.1186/1471-2466-14-32. BMC Pulm Med. 2014. PMID: 24581246 Free PMC article.
References
-
- Brook I, Gober AE. Recovery of potential pathogens and interfering bacteria in the nasopharynx of smokers and nonsmokers. Chest 2005;127:2072–2075. - PubMed
-
- Nuorti JP, Butler JC, Farley MM, Harrison LH, McGeer A, Kolczak MS, Breiman RF. Cigarette smoking and invasive pneumococcal disease: Active Bacterial Core Surveillance Team. N Engl J Med 2000;342:681–689. - PubMed
-
- US Department of Health and Human Services. The health consequences of involuntary exposure to tobacco smoke: a report of the surgeon general. Atlanta: Department of Health and Human Services, Centers for Disease Control and Prevention, Coordinating Center for Health Promotion, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; 2006.
-
- Sopori M. Effects of cigarette smoke on the immune system. Nat Rev Immunol 2002;2:372–377. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
