

Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS)
- PMID: 20352044
- PMCID: PMC2844426
- DOI: 10.1371/journal.pone.0009872
Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS)
Abstract
Background: Amyotrophic lateral sclerosis (ALS), a common late-onset neurodegenerative disease, is associated with fronto-temporal dementia (FTD) in 3-10% of patients. A mutation in CHMP2B was recently identified in a Danish pedigree with autosomal dominant FTD. Subsequently, two unrelated patients with familial ALS, one of whom also showed features of FTD, were shown to carry missense mutations in CHMP2B. The initial aim of this study was to determine whether mutations in CHMP2B contribute more broadly to ALS pathogenesis.
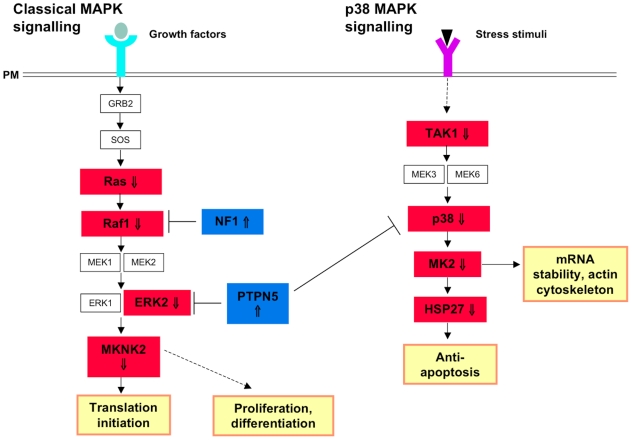
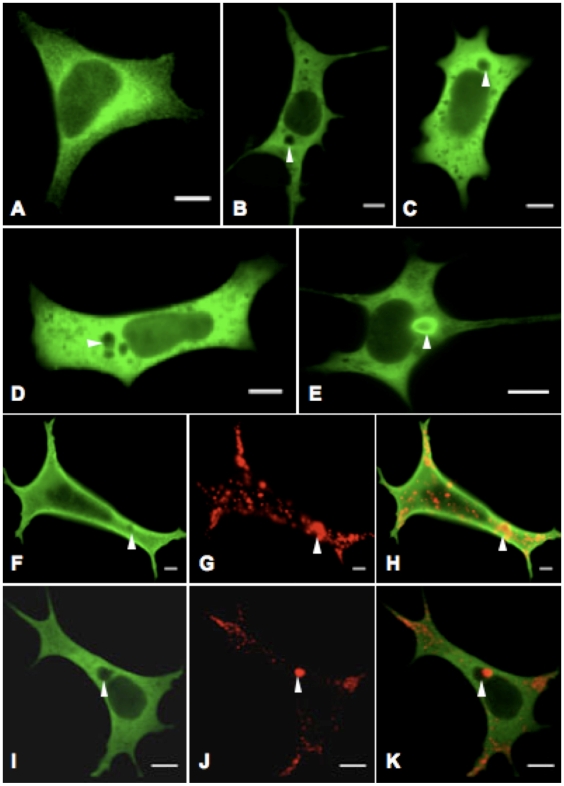
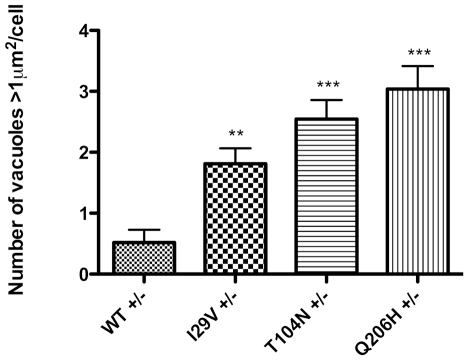
Methodology/principal findings: Sequencing of CHMP2B in 433 ALS cases from the North of England identified 4 cases carrying 3 missense mutations, including one novel mutation, p.Thr104Asn, none of which were present in 500 neurologically normal controls. Analysis of clinical and neuropathological data of these 4 cases showed a phenotype consistent with the lower motor neuron predominant (progressive muscular atrophy (PMA)) variant of ALS. Only one had a recognised family history of ALS and none had clinically apparent dementia. Microarray analysis of motor neurons from CHMP2B cases, compared to controls, showed a distinct gene expression signature with significant differential expression predicting disassembly of cell structure; increased calcium concentration in the ER lumen; decrease in the availability of ATP; down-regulation of the classical and p38 MAPK signalling pathways, reduction in autophagy initiation and a global repression of translation. Transfection of mutant CHMP2B into HEK-293 and COS-7 cells resulted in the formation of large cytoplasmic vacuoles, aberrant lysosomal localisation demonstrated by CD63 staining and impairment of autophagy indicated by increased levels of LC3-II protein. These changes were absent in control cells transfected with wild-type CHMP2B.
Conclusions/significance: We conclude that in a population drawn from North of England pathogenic CHMP2B mutations are found in approximately 1% of cases of ALS and 10% of those with lower motor neuron predominant ALS. We provide a body of evidence indicating the likely pathogenicity of the reported gene alterations. However, absolute confirmation of pathogenicity requires further evidence, including documentation of familial transmission in ALS pedigrees which might be most fruitfully explored in cases with a LMN predominant phenotype.
Conflict of interest statement
Figures





References
-
- Parkinson N, Ince PG, Smith MO, Highley R, Skibinski G, et al. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology. 2006;67:1074–1077. - PubMed
-
- Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet. 2005;37:806–808. - PubMed
-
- Babst M, Katzmann DJ, Estepa-Sabal EJ, Meerloo T, Emr SD. Escrt-III: an endosome-associated heterooligomeric protein complex required for mvb sorting. Dev Cell. 2002;3:271–282. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
