

Transient tumor-fibroblast interactions increase tumor cell malignancy by a TGF-Beta mediated mechanism in a mouse xenograft model of breast cancer
- PMID: 20352126
- PMCID: PMC2843748
- DOI: 10.1371/journal.pone.0009832
Transient tumor-fibroblast interactions increase tumor cell malignancy by a TGF-Beta mediated mechanism in a mouse xenograft model of breast cancer
Abstract
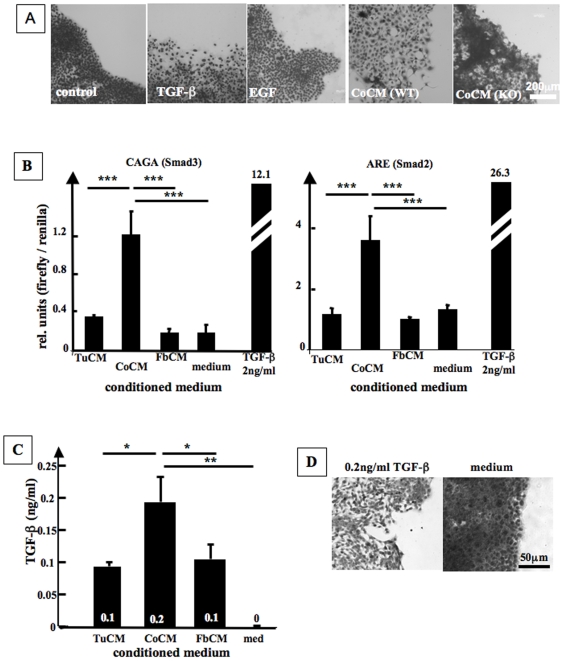
Carcinoma are complex societies of mutually interacting cells in which there is a progressive failure of normal homeostatic mechanisms, causing the parenchymal component to expand inappropriately and ultimately to disseminate to distant sites. When a cancer cell metastasizes, it first will be exposed to cancer associated fibroblasts in the immediate tumor microenvironment and then to normal fibroblasts as it traverses the underlying connective tissue towards the bloodstream. The interaction of tumor cells with stromal fibroblasts influences tumor biology by mechanisms that are not yet fully understood. Here, we report a role for normal stroma fibroblasts in the progression of invasive tumors to metastatic tumors. Using a coculture system of human metastatic breast cancer cells (MCF10CA1a) and normal murine dermal fibroblasts, we found that medium conditioned by cocultures of the two cell types (CoCM) increased migration and scattering of MCF10CA1a cells in vitro, whereas medium conditioned by homotypic cultures had little effect. Transient treatment of MCF10CA1a cells with CoCM in vitro accelerated tumor growth at orthotopic sites in vivo, and resulted in an expanded pattern of metastatic engraftment. The effects of CoCM on MCF10CA1a cells were dependent on small amounts of active TGF-beta1 secreted by fibroblasts under the influence of the tumor cells, and required intact ALK5-, p38-, and JNK signaling in the tumor cells. In conclusion, these results demonstrate that transient interactions between tumor cells and normal fibroblasts can modify the acellular component of the local microenvironment such that it induces long-lasting increases in tumorigenicity and alters the metastatic pattern of the cancer cells in vivo. TGF-beta appears to be a key player in this process, providing further rationale for the development of anti-cancer therapeutics that target the TGF-beta pathway.
Conflict of interest statement
Figures






Similar articles
-
Inhibition of TGF-β/Smad signaling by BAMBI blocks differentiation of human mesenchymal stem cells to carcinoma-associated fibroblasts and abolishes their protumor effects.Stem Cells. 2012 Dec;30(12):2810-9. doi: 10.1002/stem.1251. Stem Cells. 2012. PMID: 23034983
-
Fibroblasts direct differentiation of human breast epithelial progenitors.Breast Cancer Res. 2020 Sep 29;22(1):102. doi: 10.1186/s13058-020-01344-0. Breast Cancer Res. 2020. PMID: 32993755 Free PMC article.
-
H-Ras activation and fibroblast-induced TGF-β signaling promote laminin-332 accumulation and invasion in cutaneous squamous cell carcinoma.Matrix Biol. 2020 May;87:26-47. doi: 10.1016/j.matbio.2019.09.001. Epub 2019 Oct 24. Matrix Biol. 2020. PMID: 31655292
-
Breast cancer cells induce stromal fibroblasts to express MMP-9 via secretion of TNF-alpha and TGF-beta.J Cell Sci. 2005 May 15;118(Pt 10):2143-53. doi: 10.1242/jcs.02334. Epub 2005 Apr 26. J Cell Sci. 2005. PMID: 15855236
-
Mesenchymal Stem Cells Induce Directional Migration of Invasive Breast Cancer Cells through TGF-β.Sci Rep. 2015 Nov 20;5:16941. doi: 10.1038/srep16941. Sci Rep. 2015. PMID: 26585689 Free PMC article.
Cited by
-
TGF-beta receptor type-2 expression in cancer-associated fibroblasts regulates breast cancer cell growth and survival and is a prognostic marker in pre-menopausal breast cancer.Oncogene. 2015 Jan 2;34(1):27-38. doi: 10.1038/onc.2013.527. Epub 2013 Dec 16. Oncogene. 2015. PMID: 24336330 Clinical Trial.
-
Comparative study and meta-analysis of meta-analysis studies for the correlation of genomic markers with early cancer detection.Hum Genomics. 2013 Jun 5;7(1):14. doi: 10.1186/1479-7364-7-14. Hum Genomics. 2013. PMID: 23738773 Free PMC article.
-
Structure-guided engineering of TGF-βs for the development of novel inhibitors and probing mechanism.Bioorg Med Chem. 2018 Oct 15;26(19):5239-5246. doi: 10.1016/j.bmc.2018.07.008. Epub 2018 Jul 7. Bioorg Med Chem. 2018. PMID: 30026042 Free PMC article. Review.
-
Influence of Fibroblasts on Mammary Gland Development, Breast Cancer Microenvironment Remodeling, and Cancer Cell Dissemination.Cancers (Basel). 2020 Jun 26;12(6):1697. doi: 10.3390/cancers12061697. Cancers (Basel). 2020. PMID: 32604738 Free PMC article. Review.
-
FogBank: a single cell segmentation across multiple cell lines and image modalities.BMC Bioinformatics. 2014 Dec 30;15(1):431. doi: 10.1186/s12859-014-0431-x. BMC Bioinformatics. 2014. PMID: 25547324 Free PMC article.
References
-
- Beacham DA, Cukierman E. Stromagenesis: the changing face of fibroblastic microenvironments during tumor progression. SeminCancer Biol. 2005;15:329–341. - PubMed
-
- Proia DA, Kuperwasser C. Stroma: tumor agonist or antagonist. Cell Cycle. 2005;4:1022–1025. - PubMed
-
- Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature. 2001;411:375–379. - PubMed
-
- Baglole CJ, Ray DM, Bernstein SH, Feldon SE, Smith TJ, et al. More than structural cells, fibroblasts create and orchestrate the tumor microenvironment. ImmunolInvest. 2006;35:297–325. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
