

Identification of NAD interacting residues in proteins
- PMID: 20353553
- PMCID: PMC2853471
- DOI: 10.1186/1471-2105-11-160
Identification of NAD interacting residues in proteins
Abstract
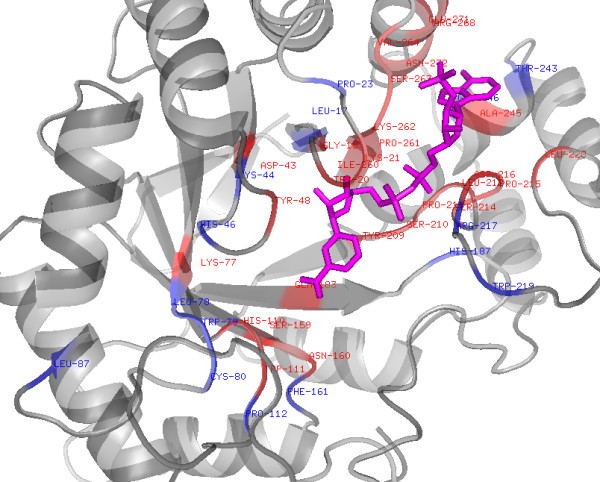
Background: Small molecular cofactors or ligands play a crucial role in the proper functioning of cells. Accurate annotation of their target proteins and binding sites is required for the complete understanding of reaction mechanisms. Nicotinamide adenine dinucleotide (NAD+ or NAD) is one of the most commonly used organic cofactors in living cells, which plays a critical role in cellular metabolism, storage and regulatory processes. In the past, several NAD binding proteins (NADBP) have been reported in the literature, which are responsible for a wide-range of activities in the cell. Attempts have been made to derive a rule for the binding of NAD+ to its target proteins. However, so far an efficient model could not be derived due to the time consuming process of structure determination, and limitations of similarity based approaches. Thus a sequence and non-similarity based method is needed to characterize the NAD binding sites to help in the annotation. In this study attempts have been made to predict NAD binding proteins and their interacting residues (NIRs) from amino acid sequence using bioinformatics tools.
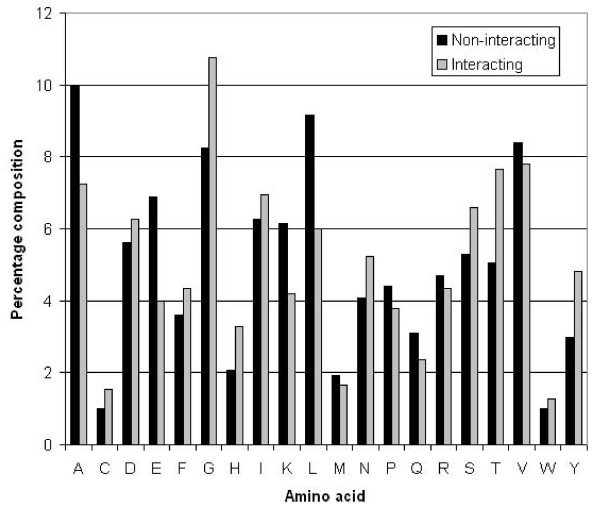
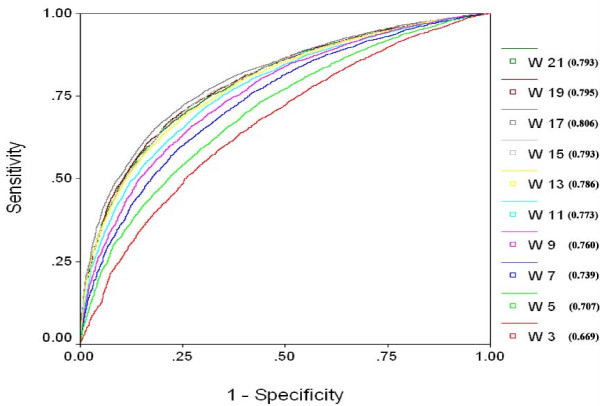
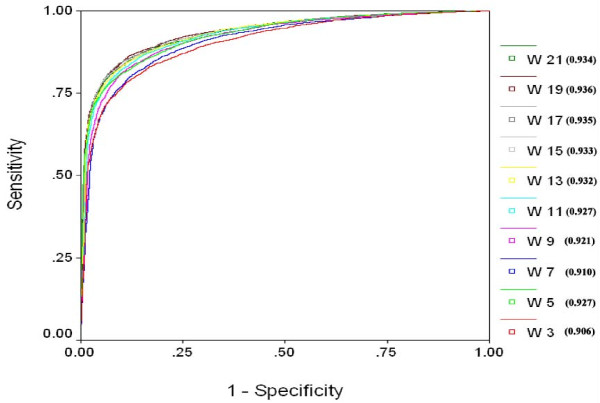
Results: We extracted 1556 proteins chains from 555 NAD binding proteins whose structure is available in Protein Data Bank. Then we removed all redundant protein chains and finally obtained 195 non-redundant NAD binding protein chains, where no two chains have more than 40% sequence identity. In this study all models were developed and evaluated using five-fold cross validation technique on the above dataset of 195 NAD binding proteins. While certain type of residues are preferred (e.g. Gly, Tyr, Thr, His) in NAD interaction, residues like Ala, Glu, Leu, Lys are not preferred. A support vector machine (SVM) based method has been developed using various window lengths of amino acid sequence for predicting NAD interacting residues and obtained maximum Matthew's correlation coefficient (MCC) 0.47 with accuracy 74.13% at window length 17. We also developed a SVM based method using evolutionary information in the form of position specific scoring matrix (PSSM) and obtained maximum MCC 0.75 with accuracy 87.25%.
Conclusion: For the first time a sequence-based method has been developed for the prediction of NAD binding proteins and their interacting residues, in the absence of any prior structural information. The present model will aid in the understanding of NAD+ dependent mechanisms of action in the cell. To provide service to the scientific community, we have developed a user-friendly web server, which is available from URL http://www.imtech.res.in/raghava/nadbinder/.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
