

trans-10,cis-12-Conjugated linoleic acid instigates inflammation in human adipocytes compared with preadipocytes
- PMID: 20353947
- PMCID: PMC2878534
- DOI: 10.1074/jbc.M109.043976
trans-10,cis-12-Conjugated linoleic acid instigates inflammation in human adipocytes compared with preadipocytes
Abstract
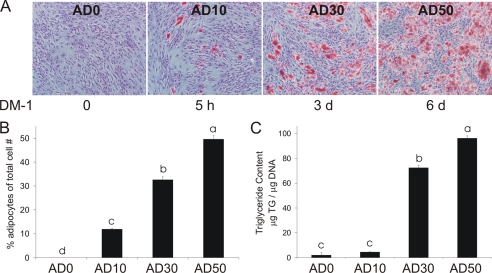
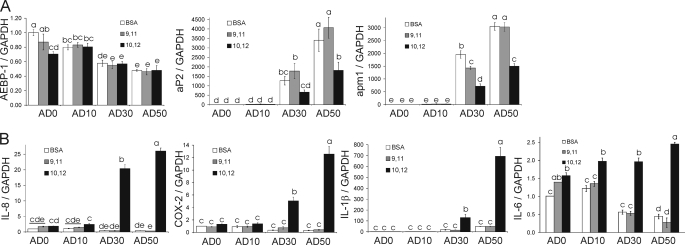
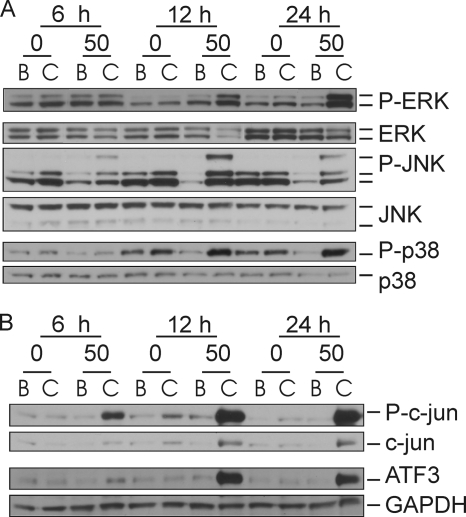
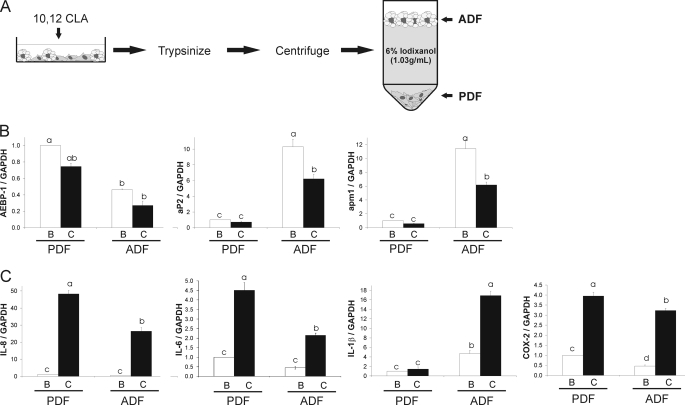
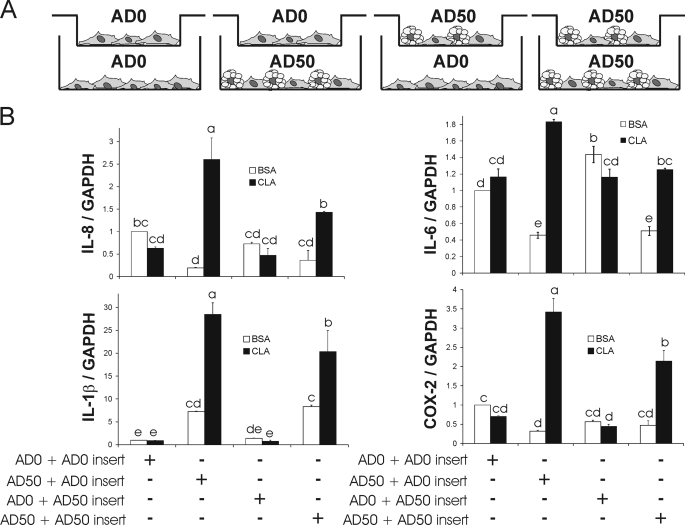
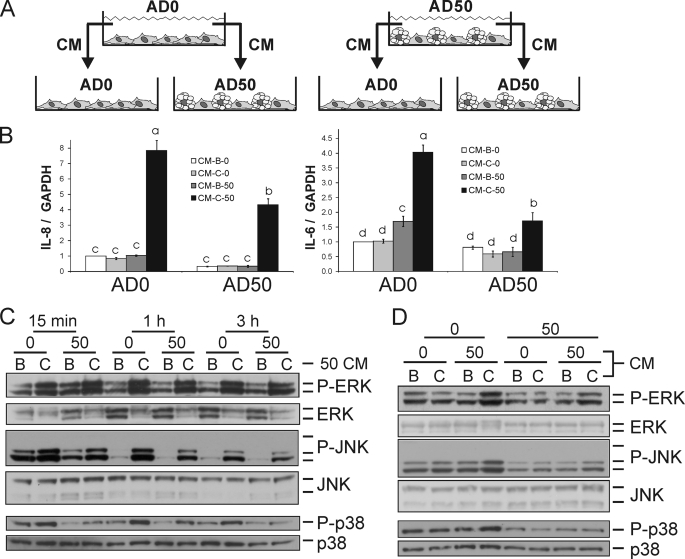
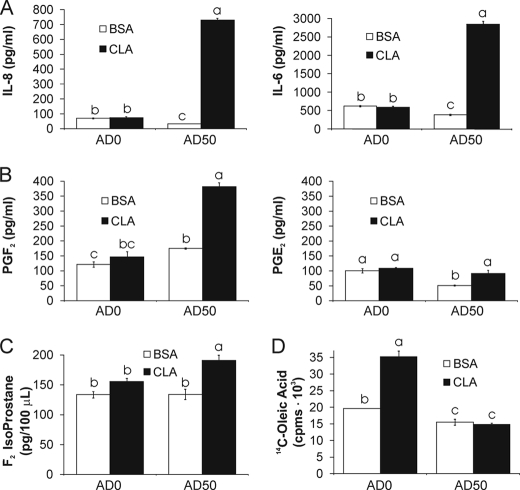
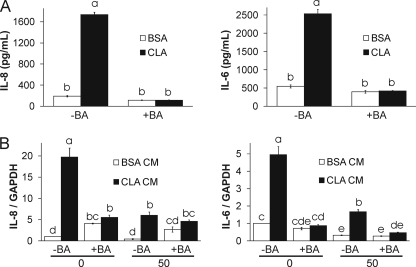
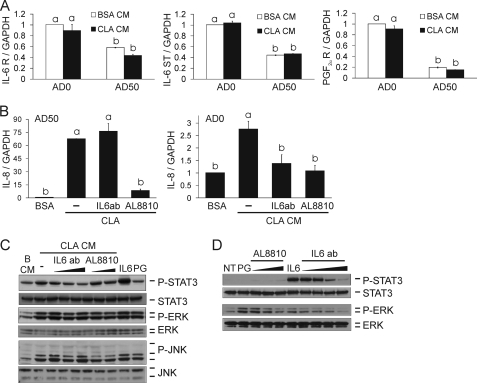
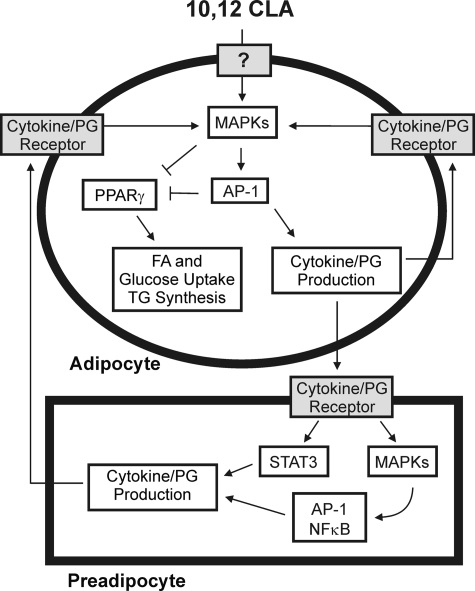
We showed previously in cultures of primary human adipocytes and preadipocytes that lipopolysaccharide and trans-10,cis-12-conjugated linoleic acid (10,12-CLA) activate the inflammatory signaling that promotes insulin resistance. Because our published data demonstrated that preadipocytes are the primary instigators of inflammatory signaling in lipopolysaccharide-treated cultures, we hypothesized that they played the same role in 10,12-CLA-mediated inflammation. To test this hypothesis, we employed four distinct models. In model 1, a differentiation model, CLA activation of MAPK and induction of interleukin-8 (IL-8), IL-6, IL-1beta, and cyclo-oxygenase-2 (COX-2) were greatest in differentiated compared with undifferentiated cultures. In model 2, a cell separation model, the mRNA levels of these inflammatory proteins were increased by 10,12-CLA compared with bovine serum albumin vehicle in the adipocyte fraction and the preadipocyte fraction. In model 3, a co-culture insert model, inserts containing approximately 50% adipocytes (AD50) or approximately 100% preadipocytes (AD0) were suspended over wells containing AD50 or AD0 cultures. 10,12-CLA-induced IL-8, IL-6, IL-1beta, and COX-2 mRNA levels were highest in AD50 cultures when co-cultured with AD0 inserts. In model 4, a conditioned medium (CM) model, CM collected from CLA-treated AD50 but not AD0 cultures induced IL-8 and IL-6 mRNA levels and activated phosphorylation of MAPK in naive AD0 and AD50 cultures. Consistent with these data, 10,12-CLA-mediated secretions of IL-8 and IL-6 from AD50 cultures were higher than from AD0 cultures. Notably, blocking adipocytokine secretion prevented the inflammatory capacity of CM from 10,12-CLA-treated cultures. These data suggest that CLA instigates the release of inflammatory signals from adipocytes that subsequently activate adjacent preadipocytes.
Figures
Similar articles
-
Conjugated linoleic acid promotes human adipocyte insulin resistance through NFkappaB-dependent cytokine production.J Biol Chem. 2005 Nov 18;280(46):38445-56. doi: 10.1074/jbc.M508159200. Epub 2005 Sep 9. J Biol Chem. 2005. PMID: 16155293 Free PMC article.
-
Inflammation and insulin resistance induced by trans-10, cis-12 conjugated linoleic acid depend on intracellular calcium levels in primary cultures of human adipocytes.J Lipid Res. 2010 Jul;51(7):1906-17. doi: 10.1194/jlr.M005447. Epub 2010 Feb 14. J Lipid Res. 2010. PMID: 20154361 Free PMC article.
-
Trans-10,cis-12 conjugated linoleic acid reduces triglyceride content while differentially affecting peroxisome proliferator activated receptor gamma2 and aP2 expression in 3T3-L1 preadipocytes.Lipids. 2001 Nov;36(11):1223-32. doi: 10.1007/s11745-001-0836-z. Lipids. 2001. PMID: 11795855
-
Conjugated linoleic acid isomers: differences in metabolism and biological effects.Biofactors. 2009 Jan-Feb;35(1):105-11. doi: 10.1002/biof.13. Biofactors. 2009. PMID: 19319853 Review.
-
Conjugated linoleic acid in humans: regulation of adiposity and insulin sensitivity.J Nutr. 2003 Oct;133(10):3041-6. doi: 10.1093/jn/133.10.3041. J Nutr. 2003. PMID: 14519781 Free PMC article. Review.
Cited by
-
Conjugated linoleic acid reduces adiposity and increases markers of browning and inflammation in white adipose tissue of mice.J Lipid Res. 2013 Apr;54(4):909-22. doi: 10.1194/jlr.M030924. Epub 2013 Feb 11. J Lipid Res. 2013. PMID: 23401602 Free PMC article.
-
Oleic acid attenuates trans-10,cis-12 conjugated linoleic acid-mediated inflammatory gene expression in human adipocytes.Lipids. 2012 Nov;47(11):1043-51. doi: 10.1007/s11745-012-3711-0. Epub 2012 Sep 2. Lipids. 2012. PMID: 22941440 Free PMC article.
-
Enhancing Bone Health with Conjugated Linoleic Acid: Mechanisms, Challenges, and Innovative Strategies.Nutrients. 2025 Apr 21;17(8):1395. doi: 10.3390/nu17081395. Nutrients. 2025. PMID: 40284258 Free PMC article. Review.
-
Diacylglycerol kinase inhibitor R59022 attenuates conjugated linoleic acid-mediated inflammation in human adipocytes.J Lipid Res. 2013 Mar;54(3):662-670. doi: 10.1194/jlr.M031211. Epub 2012 Dec 22. J Lipid Res. 2013. PMID: 23264678 Free PMC article.
-
JNK inhibition by SP600125 attenuates trans-10, cis-12 conjugated linoleic acid-mediated regulation of inflammatory and lipogenic gene expression.Lipids. 2011 Oct;46(10):885-92. doi: 10.1007/s11745-011-3587-4. Epub 2011 Jul 10. Lipids. 2011. PMID: 21744278 Free PMC article.
References
-
- Khan L., Sobush K., Keener D., Goodman K., Lowry A., Kakietek J., Zaro S. (2009) MMWR Recomm. Rep. 58, 1–26 - PubMed
-
- Park Y., Storkson J. M., Albright K. J., Liu W., Pariza M. W. (1999) Lipids 34, 235–241 - PubMed
-
- Terpstra A. H., Beynen A. C., Everts H., Kocsis S., Katan M. B., Zock P. L. (2002) J. Nutr. 132, 940–945 - PubMed
-
- Bhattacharya A., Rahman M. M., Sun D., Lawrence R., Mejia W., McCarter R., O'Shea M., Fernandes G. (2005) J. Nutr. 135, 1124–1130 - PubMed
-
- Poirier H., Shapiro J. S., Kim R. J., Lazar M. A. (2006) Diabetes 55, 1634–1641 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
