

Inhibiting the ubiquitin-proteasome system leads to preferential accumulation of toxic N-terminal mutant huntingtin fragments
- PMID: 20354076
- PMCID: PMC2876889
- DOI: 10.1093/hmg/ddq127
Inhibiting the ubiquitin-proteasome system leads to preferential accumulation of toxic N-terminal mutant huntingtin fragments
Abstract
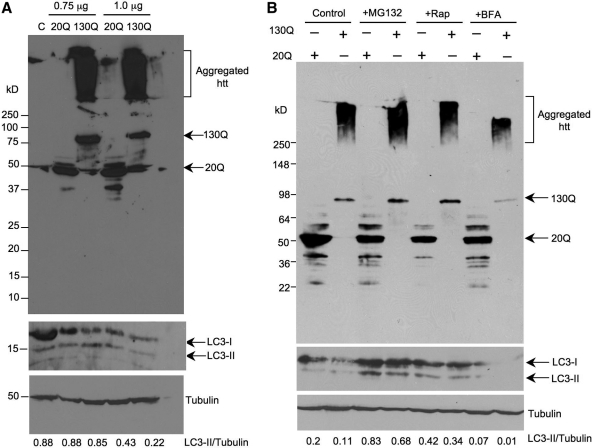
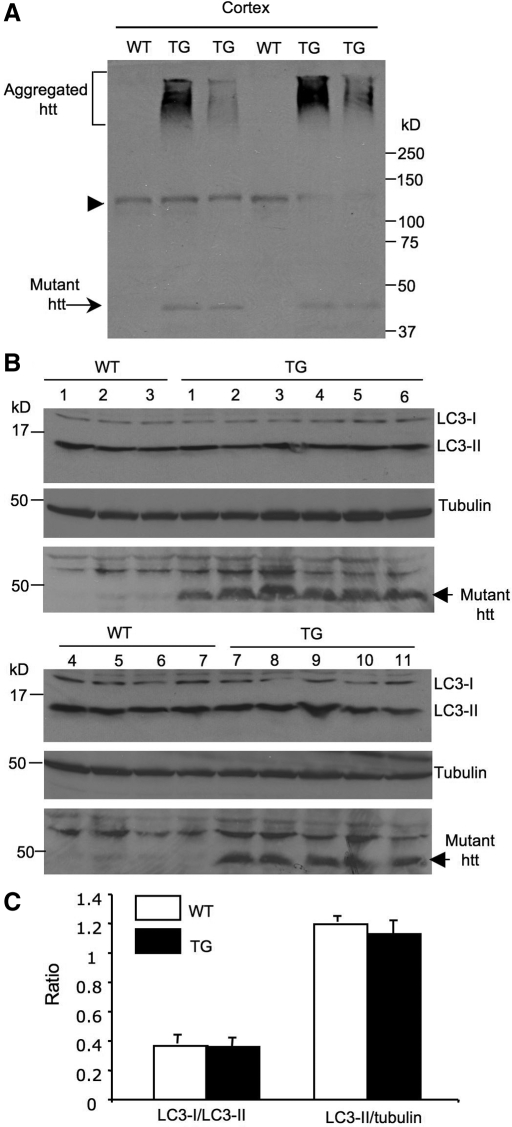
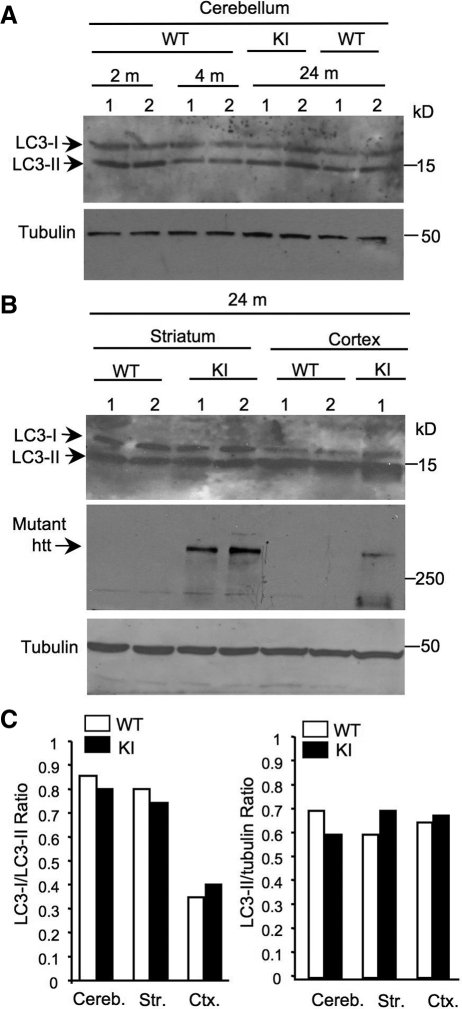
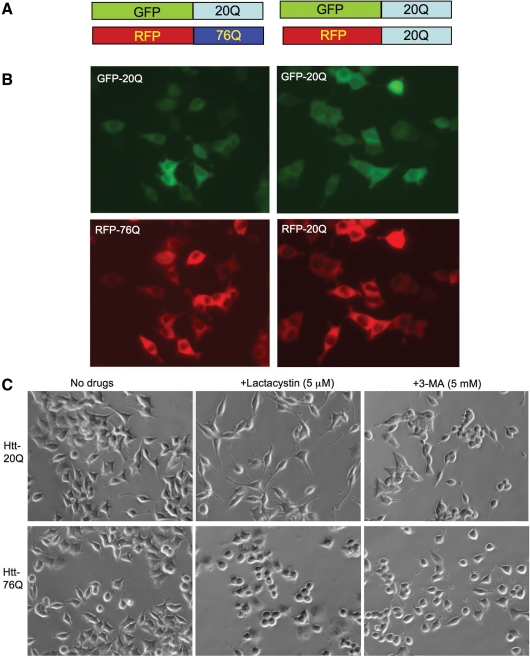
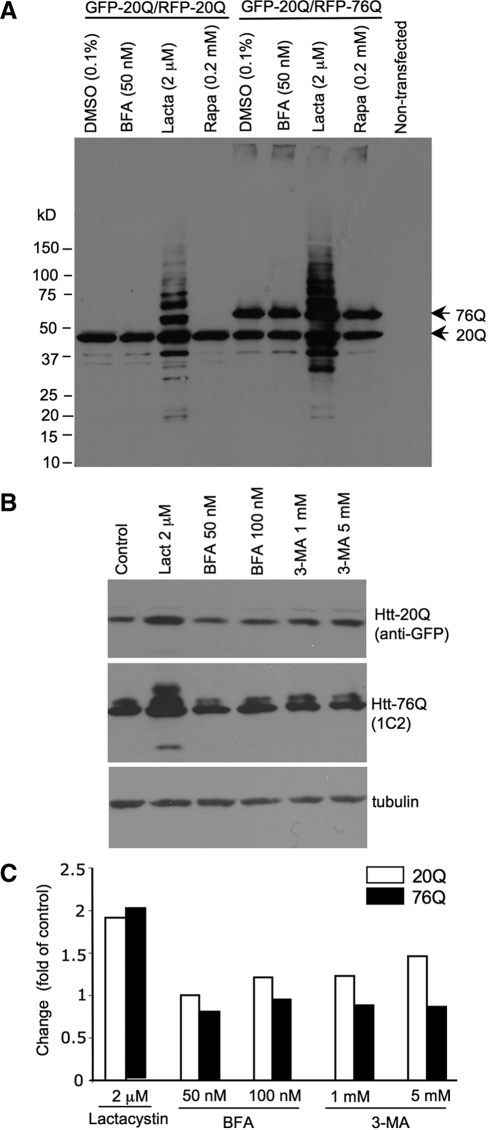
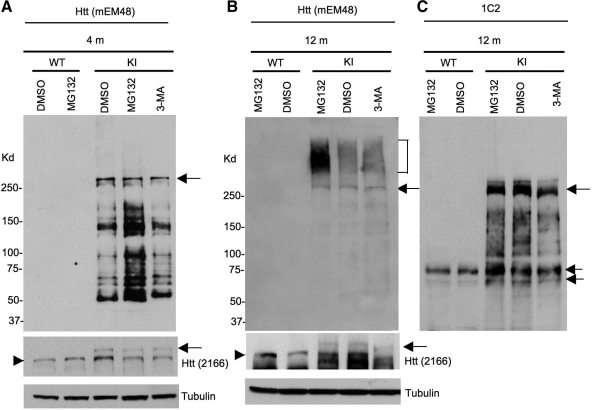
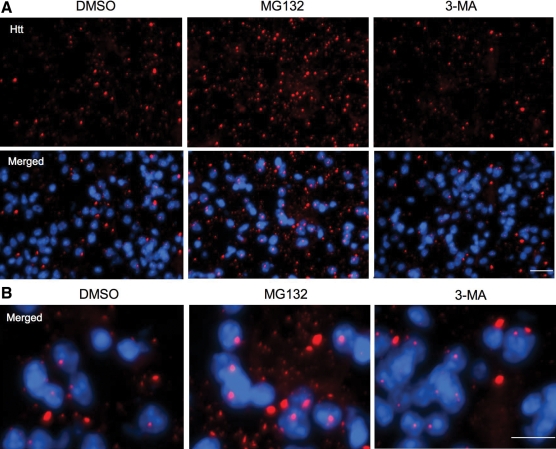
An expanded polyglutamine (polyQ) domain in the N-terminal region of huntingtin (htt) causes misfolding and accumulation of htt in neuronal cells and the subsequent neurodegeneration of Huntington's disease (HD). Clearing the misfolded htt is critical for preventing neuropathology, and this process is mediated primarily by both the ubiquitin-proteasome system (UPS) and autophagy. Although overexpression of mutant htt can inhibit UPS activity in cultured cells, mutant htt does not inhibit global UPS activity in the brains of HD transgenic mice. These findings underscore the importance of investigating the function of the UPS and autophagy in the brain when mutant proteins are not overexpressed. When cultured PC12 cells were treated with either UPS or autophagy inhibitors, more N-terminal mutant htt fragments accumulated via inhibition of the UPS. Furthermore, in HD CAG repeat knock-in mouse brain, inhibiting the UPS also resulted in a greater accumulation of N-terminal, but not full-length, mutant htt than inhibiting autophagy did. Our findings suggest that impairment of the UPS may be more important for the accumulation of N-terminal mutant htt and might therefore make an attractive therapeutic target.
Figures
References
-
- Li X., Li H., Li X.J. Intracellular degradation of misfolded proteins in polyglutamine neurodegenerative diseases. Brain Res. Rev. 2008;59:245–252. doi:10.1016/j.brainresrev.2008.08.003. - DOI - PMC - PubMed
-
- Heng M.Y., Detloff P.J., Albin R.L. Rodent genetic models of Huntington disease. Neurobiol. Dis. 2008;32:1–9. doi:10.1016/j.nbd.2008.06.005. - DOI - PubMed
-
- Ratovitski T., Nakamura M., D'Ambola J., Chighladze E., Liang Y., Wang W., Graham R., Hayden M.R., Borchelt D.R., Hirschhorn R.R., et al. N-terminal proteolysis of full-length mutant huntingtin in an inducible PC12 cell model of Huntington's disease. Cell Cycle. 2007;6:2970–2981. - PubMed
-
- Landles C., Sathasivam K., Weiss A., Woodman B., Moffitt H., Finkbeiner S., Sun B., Gafni J., Ellerby L.M., Trottier Y., et al. Proteolysis of mutant huntingtin produces an exon 1 fragment that accumulates as an aggregated protein in neuronal nuclei in Huntington's disease. J. Biol. Chem. 285:8808–8823. - PMC - PubMed
-
- DiFiglia M., Sapp E., Chase K.O., Davies S.W., Bates G.P., Vonsattel J.P., Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi:10.1126/science.277.5334.1990. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
