

Prion-like disorders: blurring the divide between transmissibility and infectivity
- PMID: 20356930
- PMCID: PMC2848109
- DOI: 10.1242/jcs.051672
Prion-like disorders: blurring the divide between transmissibility and infectivity
Abstract
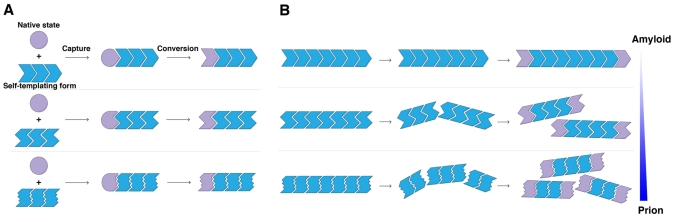
Prions are proteins that access self-templating amyloid forms, which confer phenotypic changes that can spread from individual to individual within or between species. These infectious phenotypes can be beneficial, as with yeast prions, or deleterious, as with mammalian prions that transmit spongiform encephalopathies. However, the ability to form self-templating amyloid is not unique to prion proteins. Diverse polypeptides that tend to populate intrinsically unfolded states also form self-templating amyloid conformers that are associated with devastating neurodegenerative disorders. Moreover, two RNA-binding proteins, FUS and TDP-43, which form cytoplasmic aggregates in amyotrophic lateral sclerosis, harbor a 'prion domain' similar to those found in several yeast prion proteins. Can these proteins and the neurodegenerative diseases to which they are linked become 'infectious' too? Here, we highlight advances that define the transmissibility of amyloid forms connected with Alzheimer's disease, Parkinson's disease and Huntington's disease. Collectively, these findings suggest that amyloid conformers can spread from cell to cell within the brains of afflicted individuals, thereby spreading the specific neurodegenerative phenotypes distinctive to the protein being converted to amyloid. Importantly, this transmissibility mandates a re-evaluation of emerging neuronal graft and stem-cell therapies. In this Commentary, we suggest how these treatments might be optimized to overcome the transmissible conformers that confer neurodegeneration.
Figures
References
-
- Aguzzi A. (2009). Cell biology: beyond the prion principle. Nature 459, 924-925 - PubMed
-
- Aguzzi A., Rajendran L. (2009). The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 64, 783-790 - PubMed
-
- Alper T., Cramp W. A., Haig D. A., Clarke M. C. (1967). Does the agent of scrapie replicate without nucleic acid? Nature 214, 764-766 - PubMed
-
- Auluck P. K., Chan H. Y., Trojanowski J. Q., Lee V. M., Bonini N. M. (2002). Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson's disease. Science 295, 865-868 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- DP2 OD004417/OD/NIH HHS/United States
- 1R01NS065317/NS/NINDS NIH HHS/United States
- 1R21NS067354/NS/NINDS NIH HHS/United States
- DP2 OD002177/OD/NIH HHS/United States
- R01 NS065317/NS/NINDS NIH HHS/United States
- 1DP2OD002177/OD/NIH HHS/United States
- AG00255/AG/NIA NIH HHS/United States
- T32 AG000255/AG/NIA NIH HHS/United States
- 1R01NS065317-01/NS/NINDS NIH HHS/United States
- 1DP2OD002177-01/OD/NIH HHS/United States
- 1DP2OD004417/OD/NIH HHS/United States
- 1R21NS067354-0110/NS/NINDS NIH HHS/United States
- R21 NS067354/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
