

Molecular cross talk between misfolded proteins in animal models of Alzheimer's and prion diseases
- PMID: 20357103
- PMCID: PMC2859074
- DOI: 10.1523/JNEUROSCI.5924-09.2010
Molecular cross talk between misfolded proteins in animal models of Alzheimer's and prion diseases
Abstract
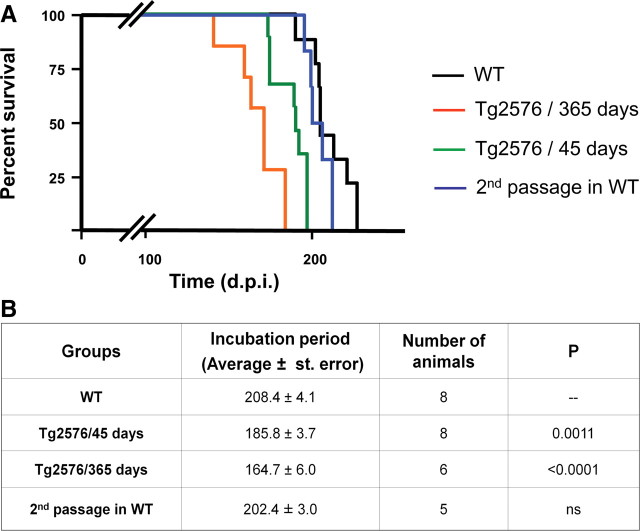
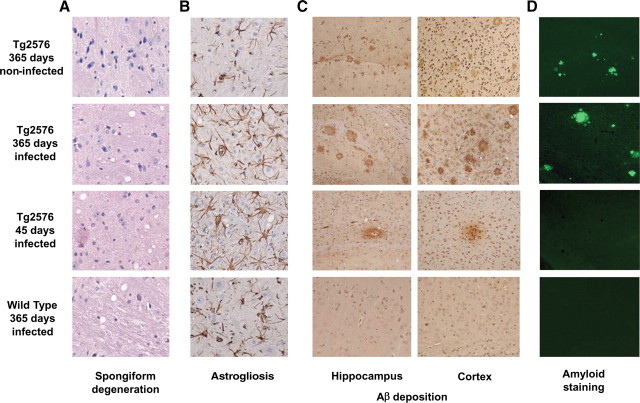
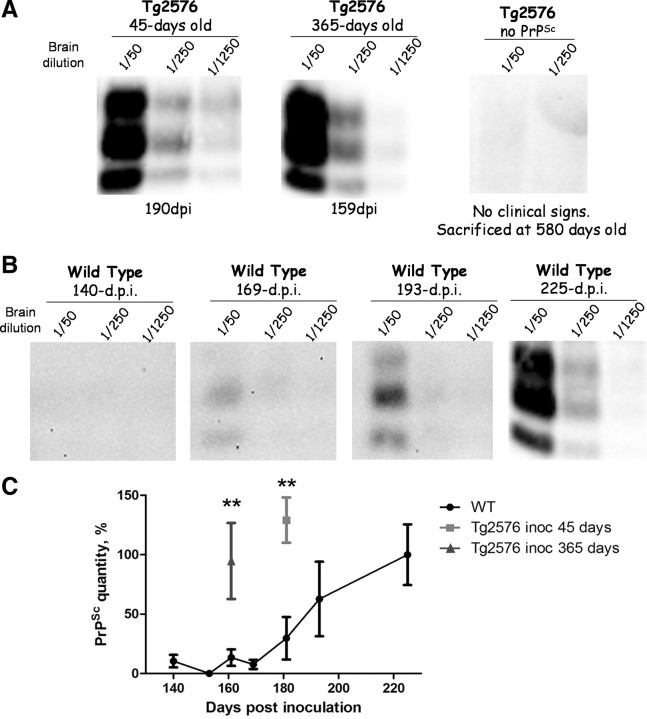
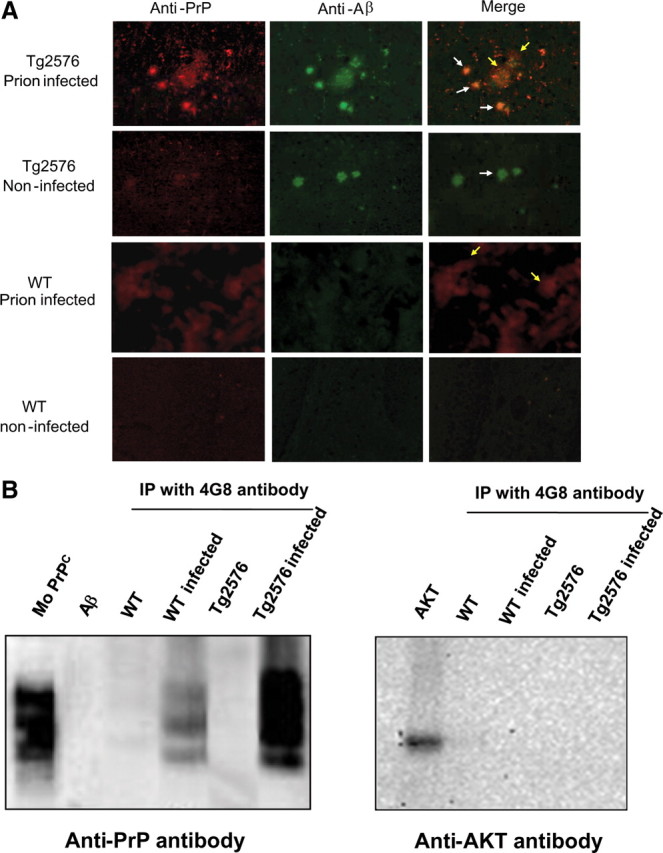
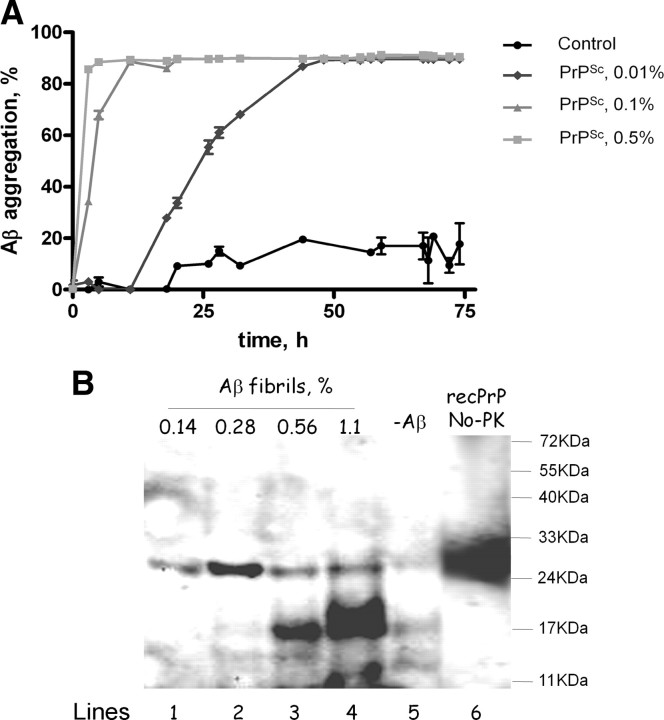
The central event in protein misfolding disorders (PMDs) is the accumulation of a misfolded form of a naturally expressed protein. Despite the diversity of clinical symptoms associated with different PMDs, many similarities in their mechanism suggest that distinct pathologies may cross talk at the molecular level. The main goal of this study was to analyze the interaction of the protein misfolding processes implicated in Alzheimer's and prion diseases. For this purpose, we inoculated prions in an Alzheimer's transgenic mouse model that develop typical amyloid plaques and followed the progression of pathological changes over time. Our findings show a dramatic acceleration and exacerbation of both pathologies. The onset of prion disease symptoms in transgenic mice appeared significantly faster with a concomitant increase on the level of misfolded prion protein in the brain. A striking increase in amyloid plaque deposition was observed in prion-infected mice compared with their noninoculated counterparts. Histological and biochemical studies showed the association of the two misfolded proteins in the brain and in vitro experiments showed that protein misfolding can be enhanced by a cross-seeding mechanism. These results suggest a profound interaction between Alzheimer's and prion pathologies, indicating that one protein misfolding process may be an important risk factor for the development of a second one. Our findings may have important implications to understand the origin and progression of PMDs.
Figures
Similar articles
-
Amyloids, prions and the inherent infectious nature of misfolded protein aggregates.Trends Biochem Sci. 2006 Mar;31(3):150-5. doi: 10.1016/j.tibs.2006.01.002. Epub 2006 Feb 13. Trends Biochem Sci. 2006. PMID: 16473510 Review.
-
Prion infection of mice transgenic for human APPSwe: increased accumulation of cortical formic acid extractable Abeta(1-42) and rapid scrapie disease development.Int J Dev Neurosci. 2008 Nov;26(7):821-4. doi: 10.1016/j.ijdevneu.2008.07.001. Epub 2008 Jul 9. Int J Dev Neurosci. 2008. PMID: 18662767
-
Anchorless prion protein results in infectious amyloid disease without clinical scrapie.Science. 2005 Jun 3;308(5727):1435-9. doi: 10.1126/science.1110837. Science. 2005. PMID: 15933194
-
Cross-seeding of prions by aggregated α-synuclein leads to transmissible spongiform encephalopathy.PLoS Pathog. 2017 Aug 10;13(8):e1006563. doi: 10.1371/journal.ppat.1006563. eCollection 2017 Aug. PLoS Pathog. 2017. PMID: 28797122 Free PMC article.
-
Prion Diseases: A Unique Transmissible Agent or a Model for Neurodegenerative Diseases?Biomolecules. 2021 Feb 2;11(2):207. doi: 10.3390/biom11020207. Biomolecules. 2021. PMID: 33540845 Free PMC article. Review.
Cited by
-
Targeting microglial K(ATP) channels to treat neurodegenerative diseases: a mitochondrial issue.Oxid Med Cell Longev. 2013;2013:194546. doi: 10.1155/2013/194546. Epub 2013 Jun 16. Oxid Med Cell Longev. 2013. PMID: 23844272 Free PMC article. Review.
-
Bacterial curli protein promotes the conversion of PAP248-286 into the amyloid SEVI: cross-seeding of dissimilar amyloid sequences.PeerJ. 2013 Feb 12;1:e5. doi: 10.7717/peerj.5. Print 2013. PeerJ. 2013. PMID: 23638387 Free PMC article.
-
Promiscuous cross-seeding between bacterial amyloids promotes interspecies biofilms.J Biol Chem. 2012 Oct 12;287(42):35092-35103. doi: 10.1074/jbc.M112.383737. Epub 2012 Aug 13. J Biol Chem. 2012. PMID: 22891247 Free PMC article.
-
Postsynaptic Receptors for Amyloid-β Oligomers as Mediators of Neuronal Damage in Alzheimer's Disease.Front Physiol. 2012 Dec 20;3:464. doi: 10.3389/fphys.2012.00464. eCollection 2012. Front Physiol. 2012. PMID: 23267328 Free PMC article.
-
Microbiome Impact on Amyloidogenesis.Front Mol Biosci. 2022 Jun 16;9:926702. doi: 10.3389/fmolb.2022.926702. eCollection 2022. Front Mol Biosci. 2022. PMID: 35782871 Free PMC article. Review.
References
-
- Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol. 2004;61:661–666. - PubMed
-
- Atarashi R, Wilham JM, Christensen L, Hughson AG, Moore RA, Johnson LM, Onwubiko HA, Priola SA, Caughey B. Simplified ultrasensitive prion detection by recombinant PrP conversion with shaking. Nat Methods. 2008;5:211–212. - PubMed
-
- Bradley R, Liberski PP. Bovine spongiform encephalopathy (BSE): the end of the beginning or the beginning of the end? Folia Neuropathol. 2004;42(Suppl A):55–68. - PubMed
-
- Brown DF, Dababo MA, Bigio EH, Risser RC, Eagan KP, Hladik CL, White CL., 3rd Neuropathologic evidence that the Lewy body variant of Alzheimer disease represents coexistence of Alzheimer disease and idiopathic Parkinson disease. J Neuropathol Exp Neurol. 1998;57:39–46. - PubMed
-
- Brown P, Jannotta F, Gibbs CJ, Jr, Baron H, Guiroy DC, Gajdusek DC. Coexistence of Creutzfeldt-Jakob disease and Alzheimer's disease in the same patient. Neurology. 1990;40:226–228. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases