

Deep mRNA sequencing for in vivo functional analysis of cardiac transcriptional regulators: application to Galphaq
- PMID: 20360248
- PMCID: PMC2891025
- DOI: 10.1161/CIRCRESAHA.110.217513
Deep mRNA sequencing for in vivo functional analysis of cardiac transcriptional regulators: application to Galphaq
Abstract
Rationale: Transcriptional profiling can detect subclinical heart disease and provide insight into disease etiology and functional status. Current microarray-based methods are expensive and subject to artifact.
Objective: To develop RNA sequencing methodologies using next generation massively parallel platforms for high throughput comprehensive analysis of individual mouse cardiac transcriptomes. To compare the results of sequencing- and array-based transcriptional profiling in the well-characterized Galphaq transgenic mouse hypertrophy/cardiomyopathy model.
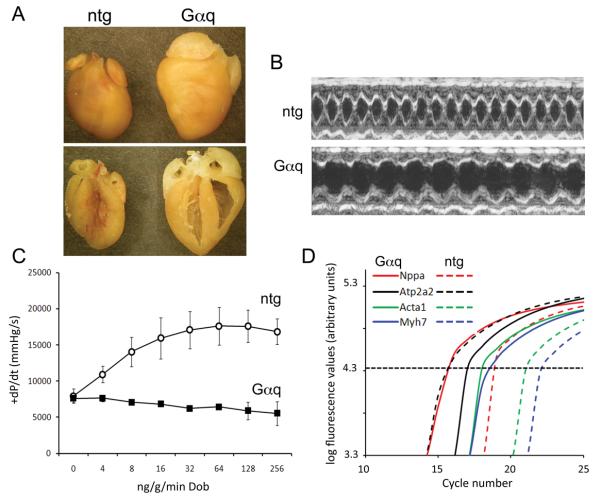
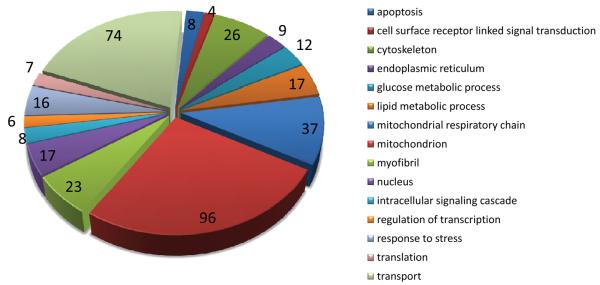
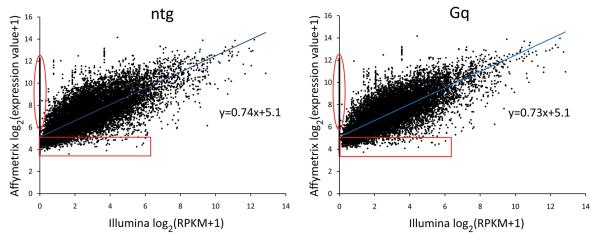
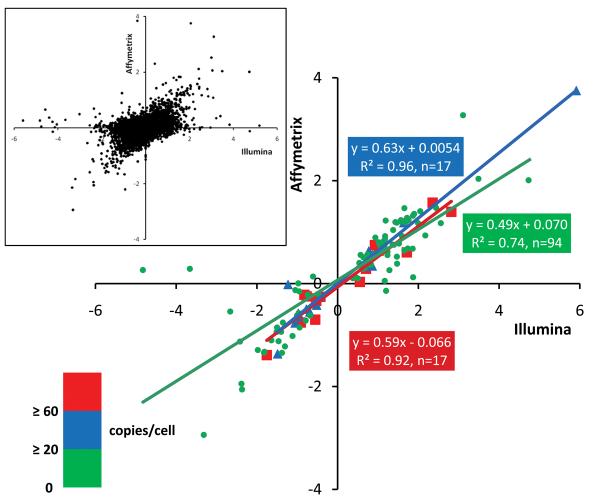
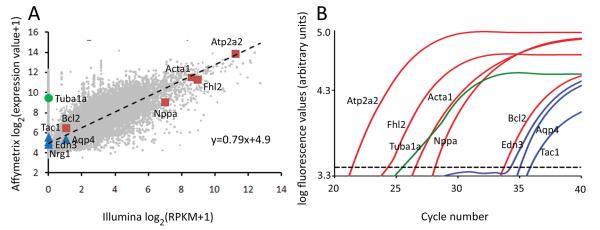
Methods and results: The techniques for preparation of individually bar-coded mouse heart RNA libraries for Illumina Genome Analyzer II resequencing are described. RNA sequencing showed that 234 high-abundance transcripts (>60 copies/cell) comprised 55% of total cardiac mRNA. Parallel transcriptional profiling of Galphaq transgenic and nontransgenic hearts by Illumina RNA sequencing and Affymetrix Mouse Gene 1.0 ST arrays revealed superior dynamic range for mRNA expression and enhanced specificity for reporting low-abundance transcripts by RNA sequencing. Differential mRNA expression in Galphaq and nontransgenic hearts correlated well between microarrays and RNA sequencing for highly abundant transcripts. RNA sequencing was superior to arrays for accurately quantifying lower-abundance genes, which represented the majority of the regulated genes in the Galphaq transgenic model.
Conclusions: RNA sequencing is rapid, accurate, and sensitive for identifying both abundant and rare cardiac transcripts, and has significant advantages in time- and cost-efficiencies over microarray analysis.
Figures
References
-
- Dorn GW, II, Robbins J, Sugden PH. Phenotyping hypertrophy - eschew obfuscation. Circ Res. 2003;92:1171–1175. - PubMed
-
- Dorn GW., II The fuzzy logic of physiological cardiac hypertrophy. Hypertension. 2007;49:962–970. - PubMed
-
- Dorn GW, II, Matkovich SJ. Put your chips on transcriptomics. Circulation. 2008;118:216–218. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
