

Systematic comparison of microarray profiling, real-time PCR, and next-generation sequencing technologies for measuring differential microRNA expression
- PMID: 20360395
- PMCID: PMC2856892
- DOI: 10.1261/rna.1947110
Systematic comparison of microarray profiling, real-time PCR, and next-generation sequencing technologies for measuring differential microRNA expression
Abstract
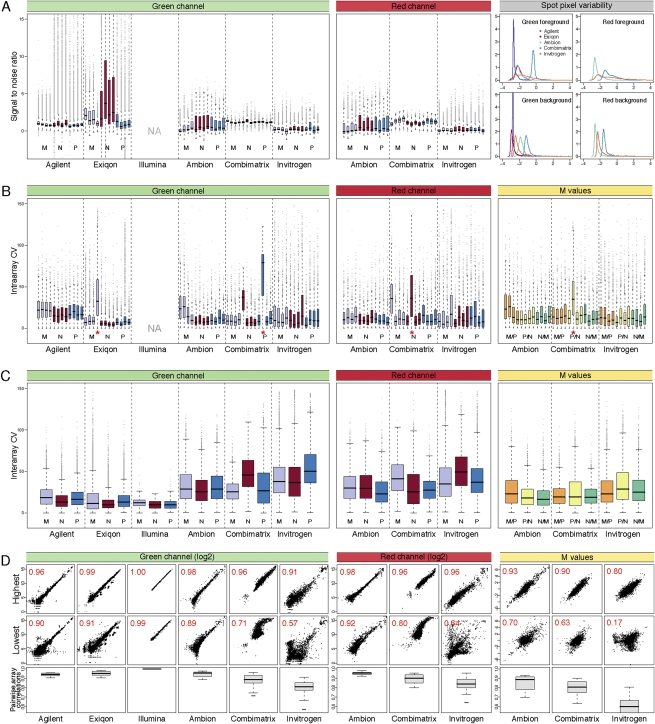
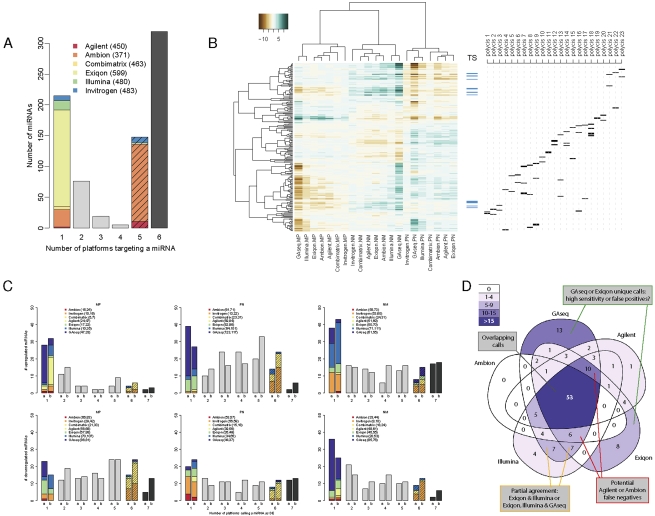
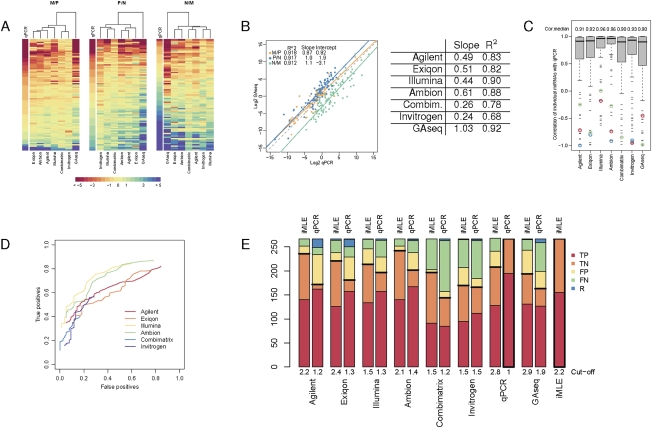
RNA abundance and DNA copy number are routinely measured in high-throughput using microarray and next-generation sequencing (NGS) technologies, and the attributes of different platforms have been extensively analyzed. Recently, the application of both microarrays and NGS has expanded to include microRNAs (miRNAs), but the relative performance of these methods has not been rigorously characterized. We analyzed three biological samples across six miRNA microarray platforms and compared their hybridization performance. We examined the utility of these platforms, as well as NGS, for the detection of differentially expressed miRNAs. We then validated the results for 89 miRNAs by real-time RT-PCR and challenged the use of this assay as a "gold standard." Finally, we implemented a novel method to evaluate false-positive and false-negative rates for all methods in the absence of a reference method.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases