

SWISS MADE: Standardized WithIn Class Sum of Squares to evaluate methodologies and dataset elements
- PMID: 20360852
- PMCID: PMC2845619
- DOI: 10.1371/journal.pone.0009905
SWISS MADE: Standardized WithIn Class Sum of Squares to evaluate methodologies and dataset elements
Abstract
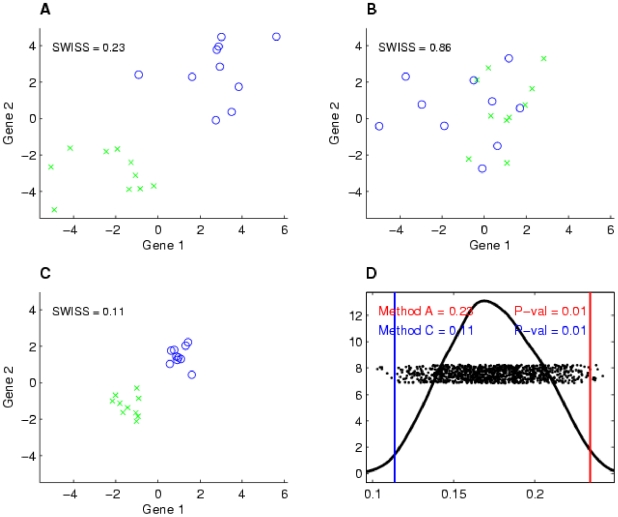
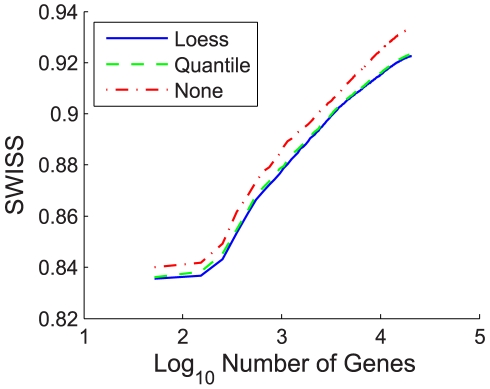
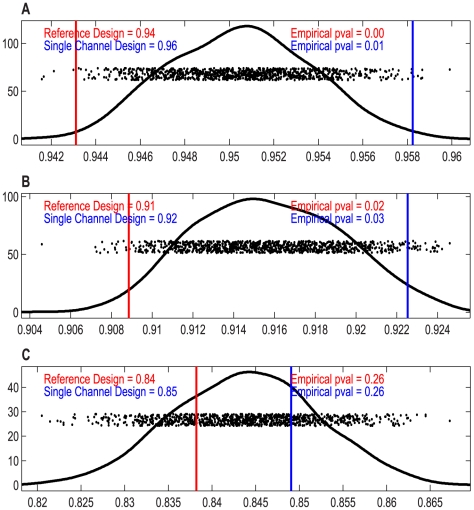
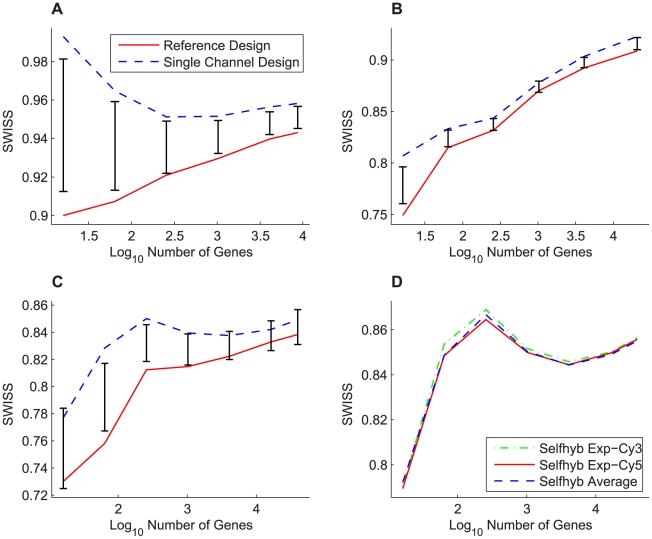
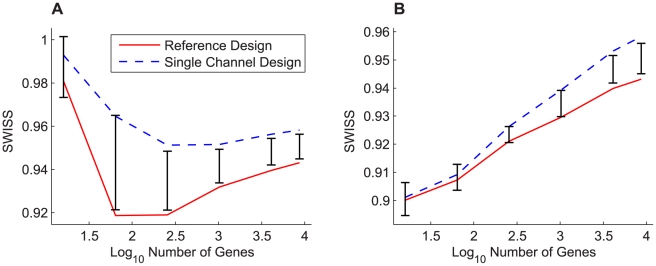
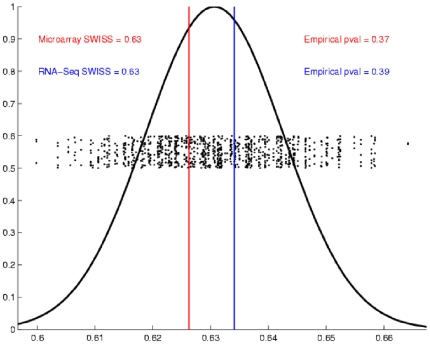
Contemporary high dimensional biological assays, such as mRNA expression microarrays, regularly involve multiple data processing steps, such as experimental processing, computational processing, sample selection, or feature selection (i.e. gene selection), prior to deriving any biological conclusions. These steps can dramatically change the interpretation of an experiment. Evaluation of processing steps has received limited attention in the literature. It is not straightforward to evaluate different processing methods and investigators are often unsure of the best method. We present a simple statistical tool, Standardized WithIn class Sum of Squares (SWISS), that allows investigators to compare alternate data processing methods, such as different experimental methods, normalizations, or technologies, on a dataset in terms of how well they cluster a priori biological classes. SWISS uses Euclidean distance to determine which method does a better job of clustering the data elements based on a priori classifications. We apply SWISS to three different gene expression applications. The first application uses four different datasets to compare different experimental methods, normalizations, and gene sets. The second application, using data from the MicroArray Quality Control (MAQC) project, compares different microarray platforms. The third application compares different technologies: a single Agilent two-color microarray versus one lane of RNA-Seq. These applications give an indication of the variety of problems that SWISS can be helpful in solving. The SWISS analysis of one-color versus two-color microarrays provides investigators who use two-color arrays the opportunity to review their results in light of a single-channel analysis, with all of the associated benefits offered by this design. Analysis of the MACQ data shows differential intersite reproducibility by array platform. SWISS also shows that one lane of RNA-Seq clusters data by biological phenotypes as well as a single Agilent two-color microarray.
Conflict of interest statement
Figures
References
-
- Bilban M, Buehler LK, Head S, Desoye G, Quaranta V. Normalizing DNA microarray data. Curr Issues Mol Biol. 2002;4:57–64. - PubMed
-
- Fan J, Ren Y. Statistical analysis of DNA microarray data in cancer research. Clin Cancer Res. 2006;12:4469–4473. - PubMed
-
- Grant GR, Manduchi E, Stoeckert CJ., Jr Analysis and management of microarray gene expression data. Curr Protoc Mol Biol Chapter. 2007;19:Unit 19 16. - PubMed
-
- Quackenbush J. Computational analysis of microarray data. Nat Rev Genet. 2001;2:418–427. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
