

Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta
- PMID: 20362275
- PMCID: PMC2850430
- DOI: 10.1016/j.ajhg.2010.02.022
Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta
Erratum in
- Am J Hum Genet. 2010 Oct 8;87(4):572-3
Abstract
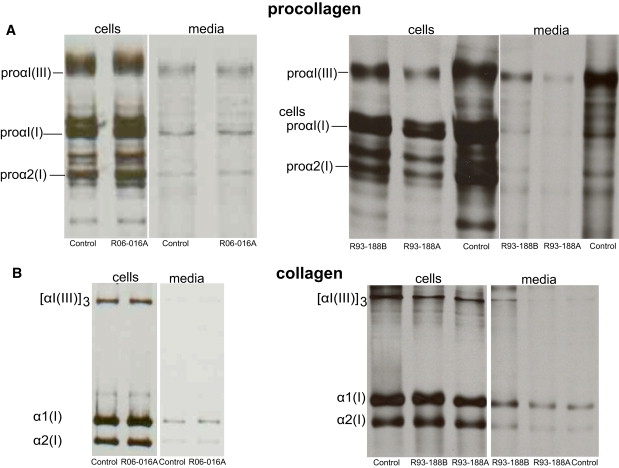
Osteogenesis imperfecta is a clinically and genetically heterogeneous brittle bone disorder that results from defects in the synthesis, structure, or posttranslational modification of type I procollagen. Dominant forms of OI result from mutations in COL1A1 or COL1A2, which encode the chains of the type I procollagen heterotrimer. The mildest form of OI typically results from diminished synthesis of structurally normal type I procollagen, whereas moderately severe to lethal forms of OI usually result from structural defects in one of the type I procollagen chains. Recessively inherited OI, usually phenotypically severe, has recently been shown to result from defects in the prolyl-3-hydroxylase complex that lead to the absence of a single 3-hydroxyproline at residue 986 of the alpha1(I) triple helical domain. We studied a cohort of five consanguineous Turkish families, originating from the Black Sea region of Turkey, with moderately severe recessively inherited OI and identified a novel locus for OI on chromosome 17. In these families, and in a Mexican-American family, homozygosity for mutations in FKBP10, which encodes FKBP65, a chaperone that participates in type I procollagen folding, was identified. Further, we determined that FKBP10 mutations affect type I procollagen secretion. These findings identify a previously unrecognized mechanism in the pathogenesis of OI.
(c) 2010 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures






Comment in
-
FKBP10 and Bruck syndrome: phenotypic heterogeneity or call for reclassification?Am J Hum Genet. 2010 Aug 13;87(2):306-7; author reply 308. doi: 10.1016/j.ajhg.2010.05.020. Am J Hum Genet. 2010. PMID: 20696291 Free PMC article. No abstract available.
References
-
- Byers, P. Disorders of Collagen Biosynthesis and Structure. In The Online Metabolic and Molecular Bases of Inherited Disease (OMMBID), D. Valle, ed. (New York: McGraw-Hill, Inc). Chapter 205, http://www.ommbid.com/.
-
- Glorieux F.H., Rauch F., Plotkin H., Ward L., Travers R., Roughley P., Lalic L., Glorieux D.F., Fassier F., Bishop N.J. Type V osteogenesis imperfecta: A new form of brittle bone disease. J. Bone Miner. Res. 2000;15:1650–1658. - PubMed
-
- Glorieux F.H., Ward L.M., Rauch F., Lalic L., Roughley P.J., Travers R. Osteogenesis imperfecta type VI: A form of brittle bone disease with a mineralization defect. J. Bone Miner. Res. 2002;17:30–38. - PubMed
-
- Morello R., Bertin T.K., Chen Y., Hicks J., Tonachini L., Monticone M., Castagnola P., Rauch F., Glorieux F.H., Vranka J. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006;127:291–304. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
