

Mutation spectrum revealed by breakpoint sequencing of human germline CNVs
- PMID: 20364136
- PMCID: PMC3428939
- DOI: 10.1038/ng.564
Mutation spectrum revealed by breakpoint sequencing of human germline CNVs
Abstract
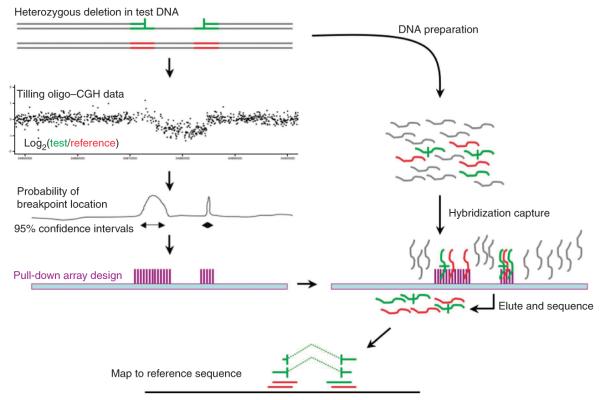
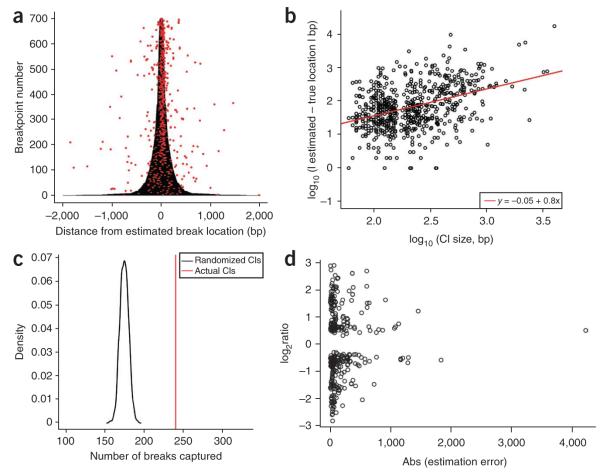
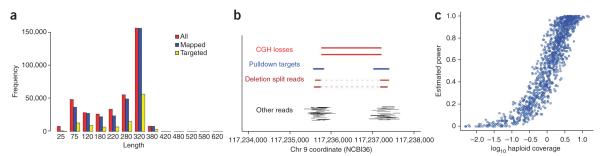
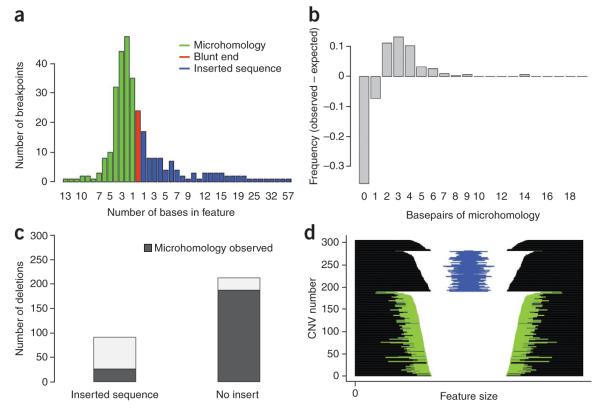
Precisely characterizing the breakpoints of copy number variants (CNVs) is crucial for assessing their functional impact. However, fewer than 10% of known germline CNVs have been mapped to the single-nucleotide level. We characterized the sequence breakpoints from a dataset of all CNVs detected in three unrelated individuals in previous array-based CNV discovery experiments. We used targeted hybridization-based DNA capture and 454 sequencing to sequence 324 CNV breakpoints, including 315 deletions. We observed two major breakpoint signatures: 70% of the deletion breakpoints have 1-30 bp of microhomology, whereas 33% of deletion breakpoints contain 1-367 bp of inserted sequence. The co-occurrence of microhomology and inserted sequence is low (10%), suggesting that there are at least two different mutational mechanisms. Approximately 5% of the breakpoints represent more complex rearrangements, including local microinversions, suggesting a replication-based strand switching mechanism. Despite a rich literature on DNA repair processes, reconstruction of the molecular events generating each of these mutations is not yet possible.
Figures
Comment in
-
Copy number variation and human genome maps.Nat Genet. 2010 May;42(5):365-6. doi: 10.1038/ng0510-365. Nat Genet. 2010. PMID: 20428091
References
-
- Wyman C, Kanaar R. DNA double-strand break repair: all’s well that ends well. Annu. Rev. Genet. 2006;40:363–383. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
