

Discovery of common Asian copy number variants using integrated high-resolution array CGH and massively parallel DNA sequencing
- PMID: 20364138
- PMCID: PMC3329635
- DOI: 10.1038/ng.555
Discovery of common Asian copy number variants using integrated high-resolution array CGH and massively parallel DNA sequencing
Abstract
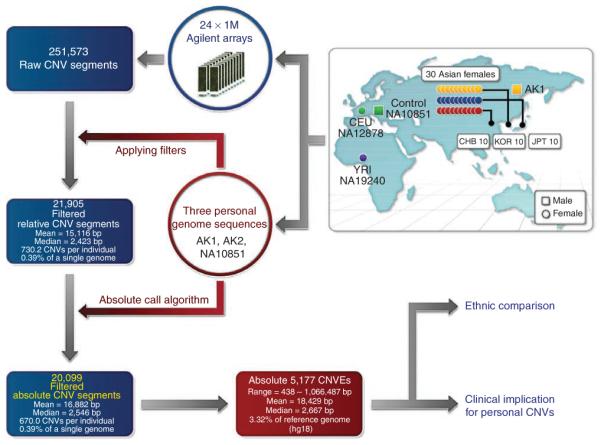
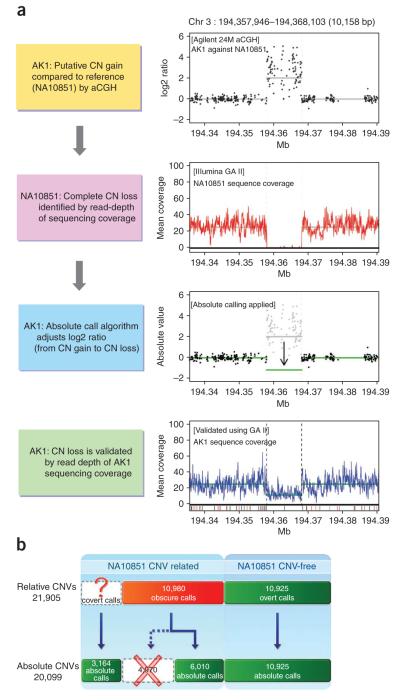
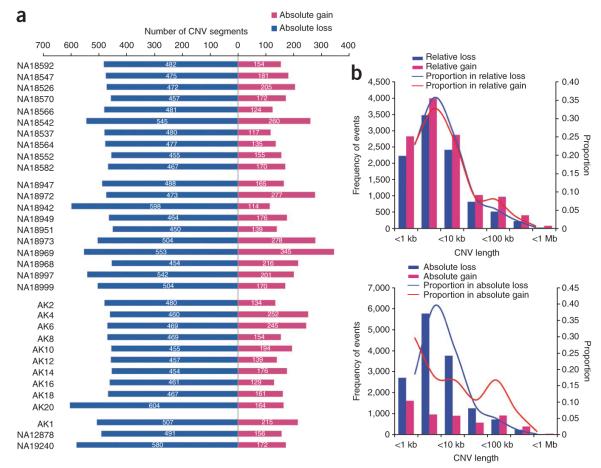
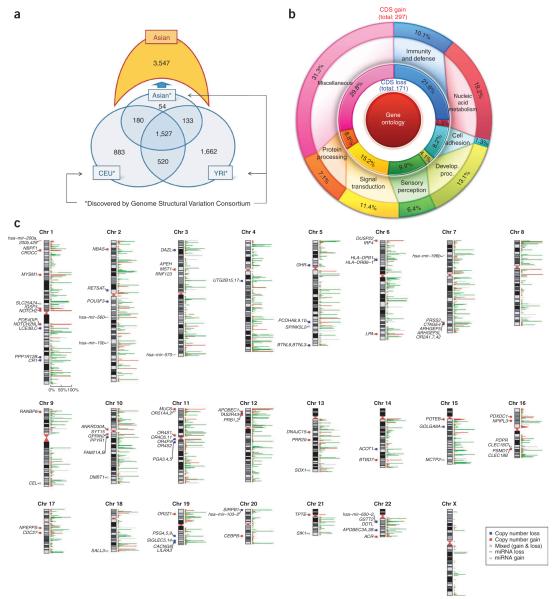
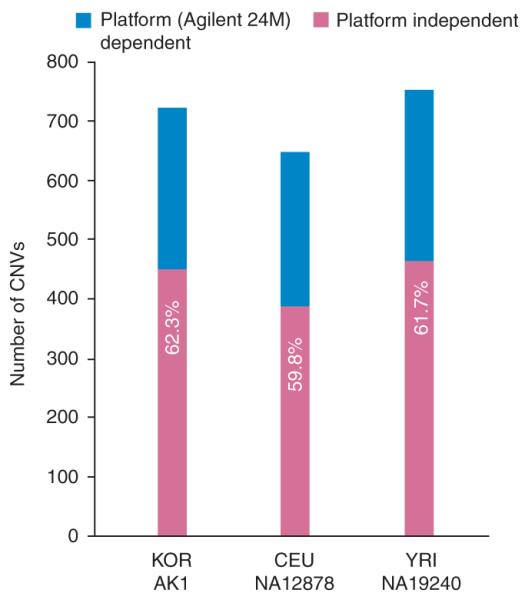
Copy number variants (CNVs) account for the majority of human genomic diversity in terms of base coverage. Here, we have developed and applied a new method to combine high-resolution array comparative genomic hybridization (CGH) data with whole-genome DNA sequencing data to obtain a comprehensive catalog of common CNVs in Asian individuals. The genomes of 30 individuals from three Asian populations (Korean, Chinese and Japanese) were interrogated with an ultra-high-resolution array CGH platform containing 24 million probes. Whole-genome sequencing data from a reference genome (NA10851, with 28.3x coverage) and two Asian genomes (AK1, with 27.8x coverage and AK2, with 32.0x coverage) were used to transform the relative copy number information obtained from array CGH experiments into absolute copy number values. We discovered 5,177 CNVs, of which 3,547 were putative Asian-specific CNVs. These common CNVs in Asian populations will be a useful resource for subsequent genetic studies in these populations, and the new method of calling absolute CNVs will be essential for applying CNV data to personalized medicine.
Figures
Comment in
-
Copy number variation and human genome maps.Nat Genet. 2010 May;42(5):365-6. doi: 10.1038/ng0510-365. Nat Genet. 2010. PMID: 20428091
References
-
- Iafrate AJ, et al. Detection of large-scale variation in the human genome. Nat. Genet. 2004;36:949–951. - PubMed
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
