

Targeting iron homeostasis induces cellular differentiation and synergizes with differentiating agents in acute myeloid leukemia
- PMID: 20368581
- PMCID: PMC2856037
- DOI: 10.1084/jem.20091488
Targeting iron homeostasis induces cellular differentiation and synergizes with differentiating agents in acute myeloid leukemia
Abstract
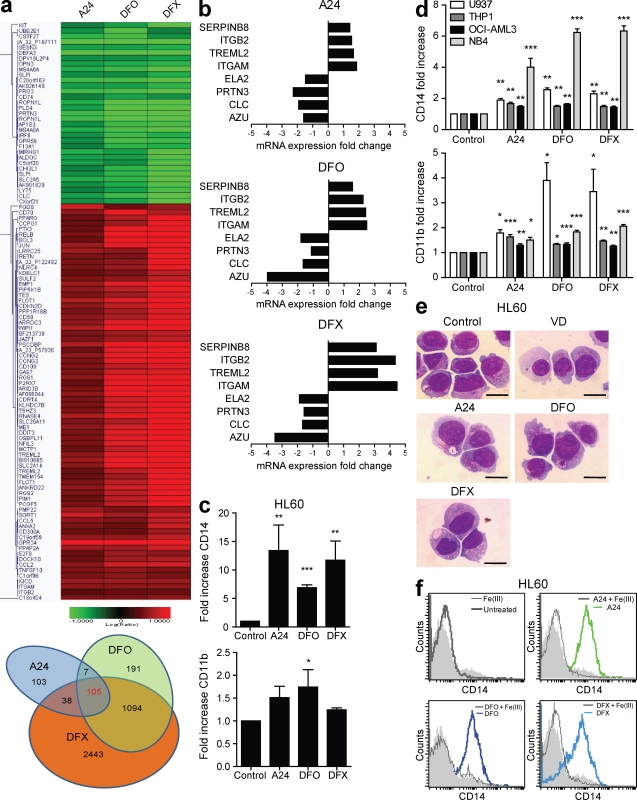
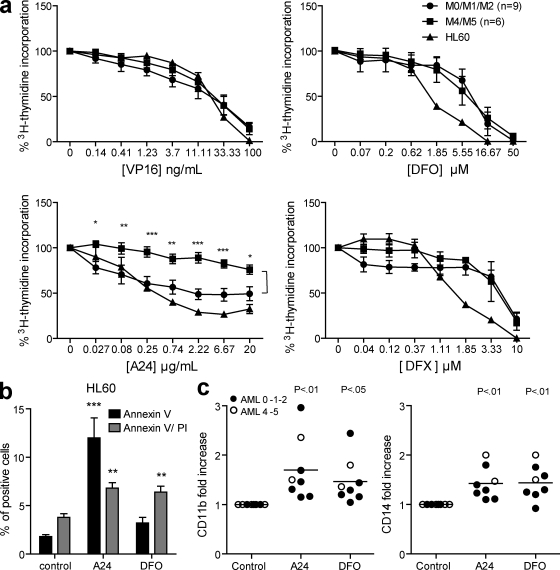
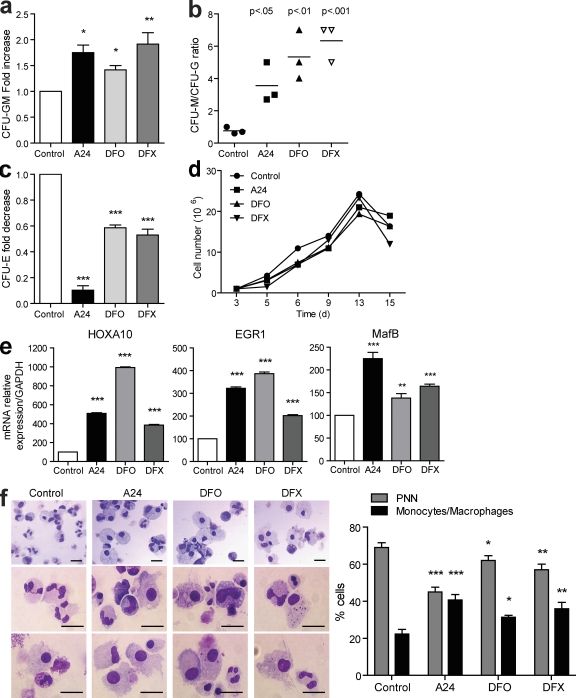
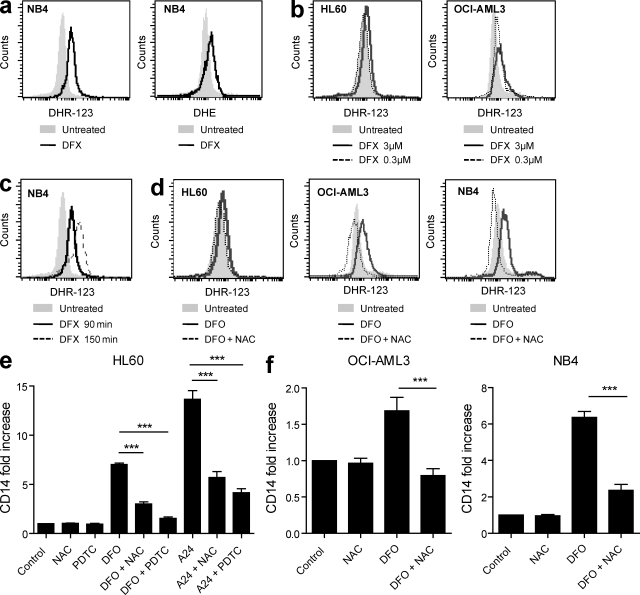
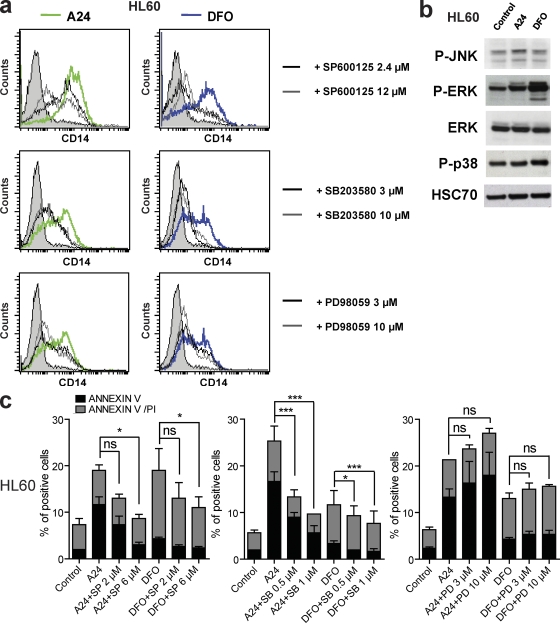
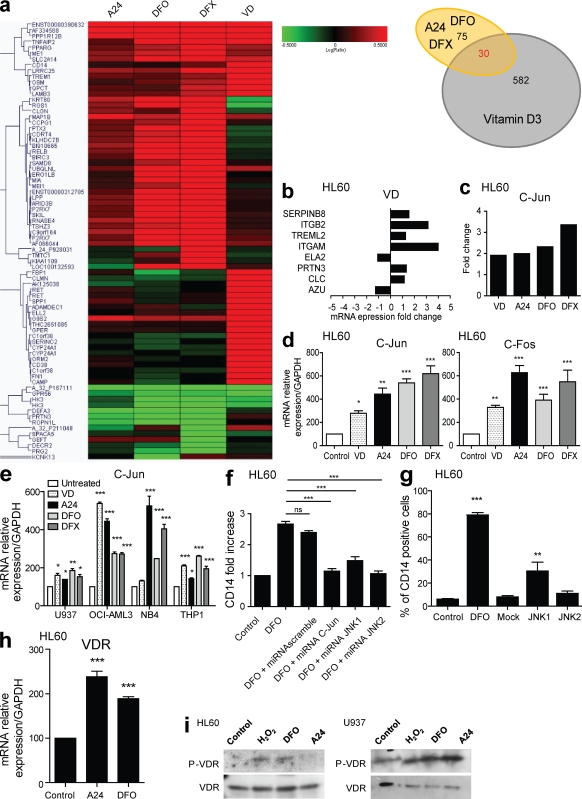
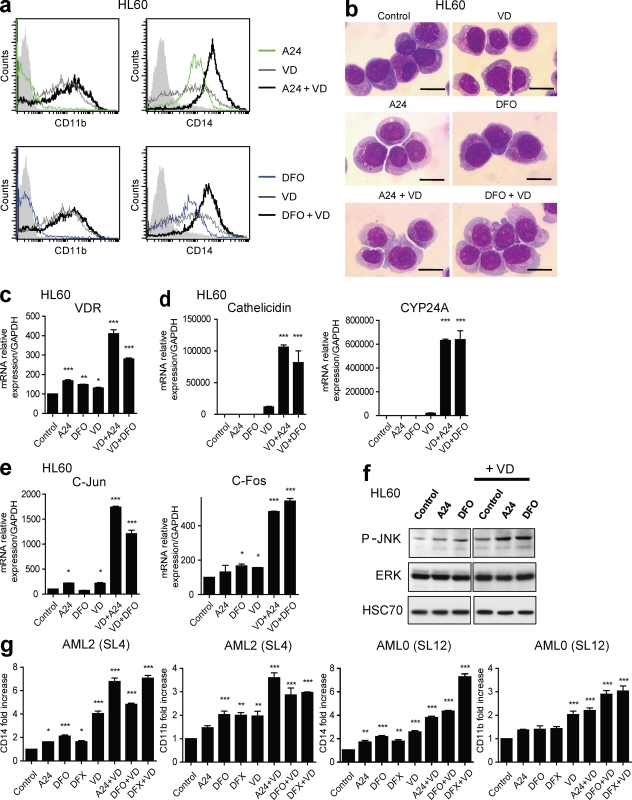
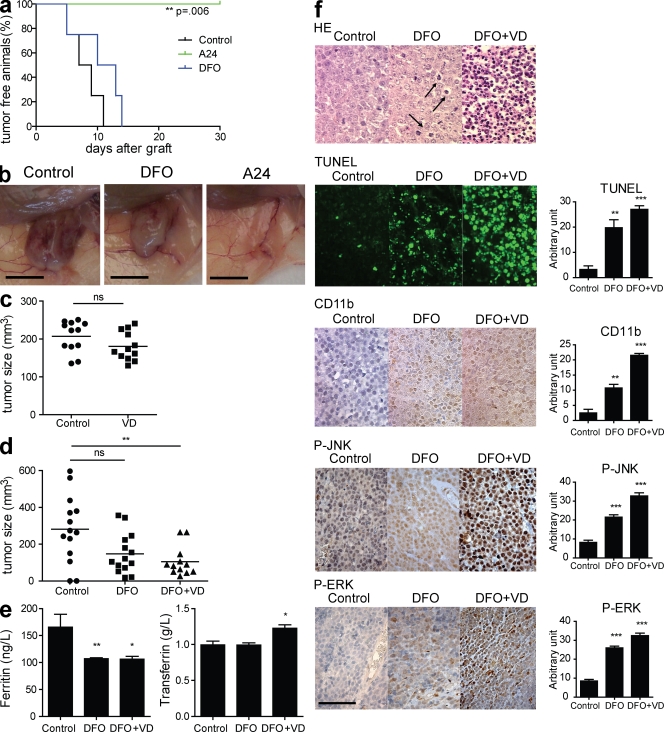
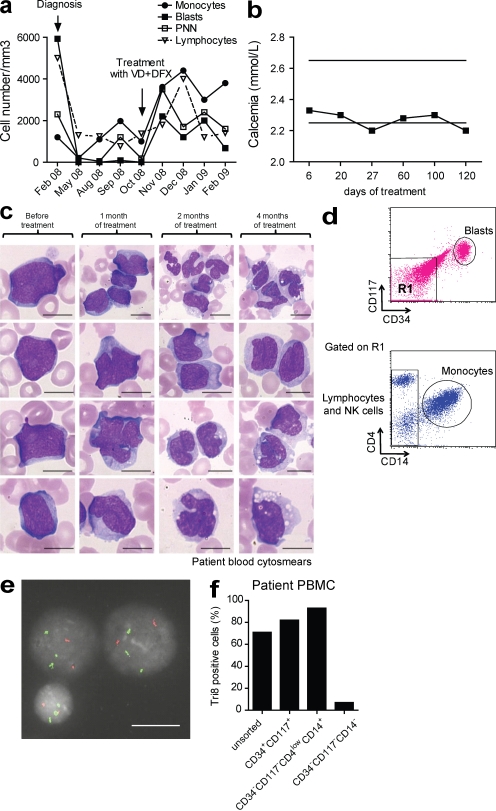
Differentiating agents have been proposed to overcome the impaired cellular differentiation in acute myeloid leukemia (AML). However, only the combinations of all-trans retinoic acid or arsenic trioxide with chemotherapy have been successful, and only in treating acute promyelocytic leukemia (also called AML3). We show that iron homeostasis is an effective target in the treatment of AML. Iron chelating therapy induces the differentiation of leukemia blasts and normal bone marrow precursors into monocytes/macrophages in a manner involving modulation of reactive oxygen species expression and the activation of mitogen-activated protein kinases (MAPKs). 30% of the genes most strongly induced by iron deprivation are also targeted by vitamin D3 (VD), a well known differentiating agent. Iron chelating agents induce expression and phosphorylation of the VD receptor (VDR), and iron deprivation and VD act synergistically. VD magnifies activation of MAPK JNK and the induction of VDR target genes. When used to treat one AML patient refractory to chemotherapy, the combination of iron-chelating agents and VD resulted in reversal of pancytopenia and in blast differentiation. We propose that iron availability modulates myeloid cell commitment and that targeting this cellular differentiation pathway together with conventional differentiating agents provides new therapeutic modalities for AML.
Figures
Comment in
-
Metabolism and the leukemic stem cell.J Exp Med. 2010 Apr 12;207(4):677-80. doi: 10.1084/jem.20100523. Epub 2010 Apr 5. J Exp Med. 2010. PMID: 20368582 Free PMC article.
References
-
- Adamson J.W. 1994. The relationship of erythropoietin and iron metabolism to red blood cell production in humans. Semin. Oncol. 21:9–15 - PubMed
-
- Bastie J.N., Balitrand N., Guidez F., Guillemot I., Larghero J., Calabresse C., Chomienne C., Delva L. 2004. 1 alpha,25-dihydroxyvitamin D3 transrepresses retinoic acid transcriptional activity via vitamin D receptor in myeloid cells. Mol. Endocrinol. 18:2685–2699 10.1210/me.2003-0412 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
