

Frontotemporal lobar degeneration: epidemiology, pathophysiology, diagnosis and management
- PMID: 20369906
- PMCID: PMC2916644
- DOI: 10.2165/11533100-000000000-00000
Frontotemporal lobar degeneration: epidemiology, pathophysiology, diagnosis and management
Abstract
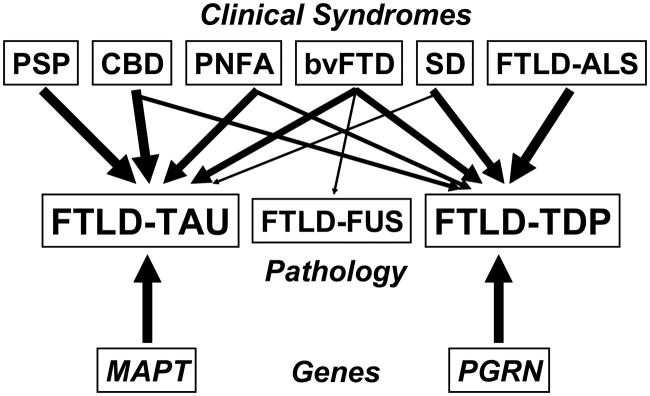
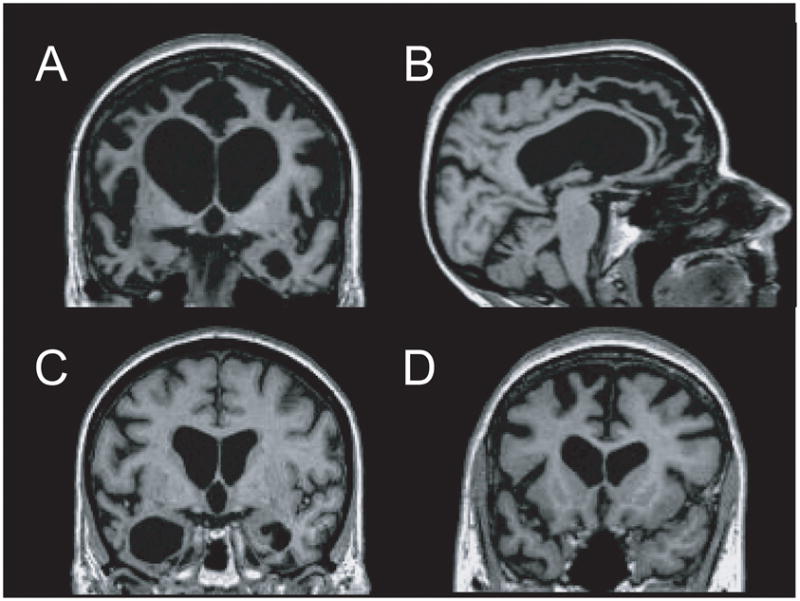
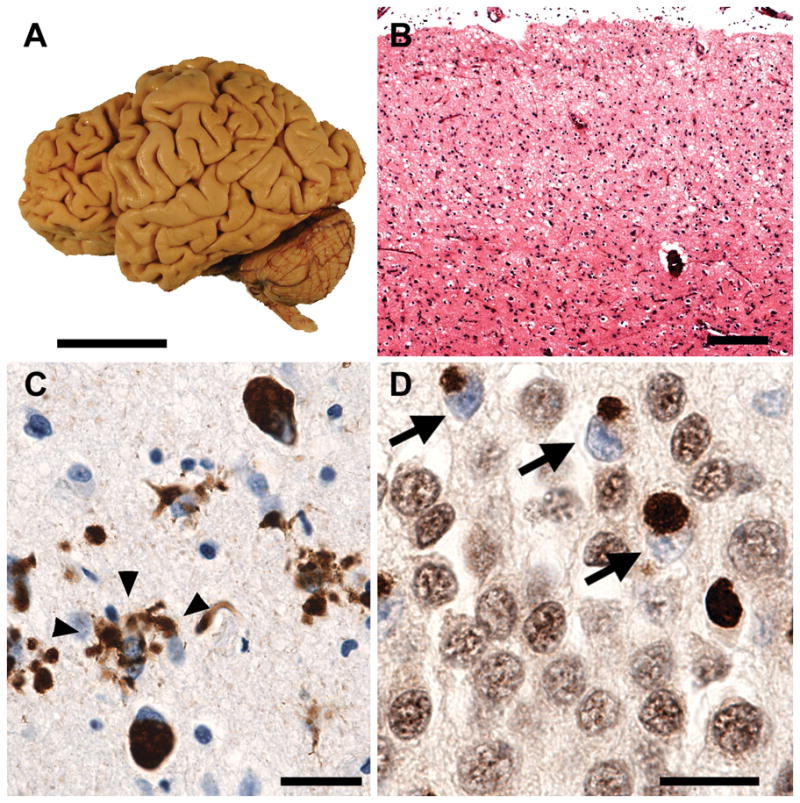
Frontotemporal lobar degeneration (FTLD) is a clinically and pathologically heterogeneous syndrome, characterized by progressive decline in behaviour or language associated with degeneration of the frontal and anterior temporal lobes. While the seminal cases were described at the turn of the 20th century, FTLD has only recently been appreciated as a leading cause of dementia, particularly in patients presenting before the age of 65 years. Three distinct clinical variants of FTLD have been described: (i) behavioural-variant frontotemporal dementia, characterized by changes in behaviour and personality in association with frontal-predominant cortical degeneration; (ii) semantic dementia, a syndrome of progressive loss of knowledge about words and objects associated with anterior temporal neuronal loss; and (iii) progressive nonfluent aphasia, characterized by effortful language output, loss of grammar and motor speech deficits in the setting of left perisylvian cortical atrophy. The majority of pathologies associated with FTLD clinical syndromes include either tau-positive (FTLD-TAU) or TAR DNA-binding protein 43 (TDP-43)-positive (FTLD-TDP) inclusion bodies. FTLD overlaps clinically and pathologically with the atypical parkinsonian disorders corticobasal degeneration and progressive supranuclear palsy, and with amyotrophic lateral sclerosis. The majority of familial FTLD cases are caused by mutations in the genes encoding microtubule-associated protein tau (leading to FTLD-TAU) or progranulin (leading to FTLD-TDP). The clinical and pathological heterogeneity of FTLD poses a significant diagnostic challenge, and in vivo prediction of underlying histopathology can be significantly improved by supplementing the clinical evaluation with genetic tests and emerging biological markers. Current pharmacotherapy for FTLD focuses on manipulating serotonergic or dopaminergic neurotransmitter systems to ameliorate behavioural or motor symptoms. However, recent advances in FTLD genetics and molecular pathology make the prospect of biologically driven, disease-specific therapies for FTLD seem closer than ever.
Conflict of interest statement
The authors have no conflicts of interest that are directly relevant to the content of this review. Dr. Rabinovici has received personal compensation for serving on scientific advisory boards for General Electric Healthcare and Novartis Diagnostics. Dr Miller has received personal compensation for participating in the speaker’s bureau for Novartis Pharmaceuticals and Pfizer.
Figures
References
-
- Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–54. - PubMed
-
- Rosso SM, Donker Kaat L, Baks T, et al. Frontotemporal dementia in the Netherlands: patient characteristics and prevalence estimates from a population-based study. Brain. 2003;126:2016–22. - PubMed
-
- Ratnavalli E, Brayne C, Dawson K, et al. The prevalence of frontotemporal dementia. Neurology. 2002;58:1615–21. - PubMed
-
- Mercy L, Hodges JR, Dawson K, et al. Incidence of early-onset dementias in Cambridgeshire, United Kingdom. Neurology. 2008;71:1496–9. - PubMed
-
- Knopman DS, Petersen RC, Edland SD, et al. The incidence of frontotemporal lobar degeneration in Rochester, Minnesota, 1990 through 1994. Neurology. 2004;62:506–8. - PubMed
New references
-
- Pick A. Uber die Beziehungen der senilen Hirnatrophie zur Aphasie. Prager Medizinische Wochenschrift. 1892;17:165–167.
-
- Pick A. Zur symptomatologie der linksseitigen schlafenappenatrophie. Monatsschrift fur Psychiatrie und Neurologie. 1904;16:378–388.
-
- Benajiba L, Le Ber I, Camuzat A, et al. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol. 2009;65(4):470–3. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
