

Phylogenetic assessment of alignments reveals neglected tree signal in gaps
- PMID: 20370897
- PMCID: PMC2884540
- DOI: 10.1186/gb-2010-11-4-r37
Phylogenetic assessment of alignments reveals neglected tree signal in gaps
Abstract
Background: The alignment of biological sequences is of chief importance to most evolutionary and comparative genomics studies, yet the two main approaches used to assess alignment accuracy have flaws: reference alignments are derived from the biased sample of proteins with known structure, and simulated data lack realism.
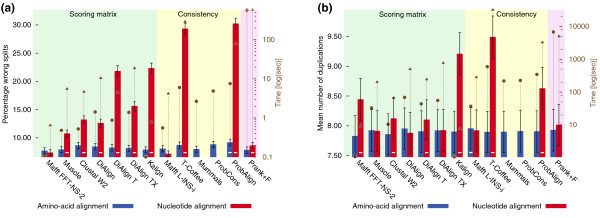
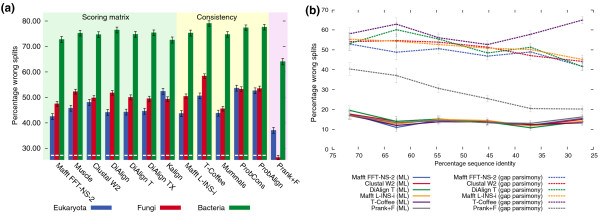
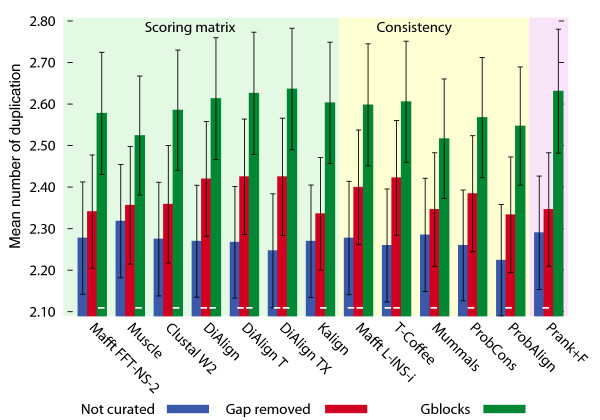
Results: Here, we introduce tree-based tests of alignment accuracy, which not only use large and representative samples of real biological data, but also enable the evaluation of the effect of gap placement on phylogenetic inference. We show that (i) the current belief that consistency-based alignments outperform scoring matrix-based alignments is misguided; (ii) gaps carry substantial phylogenetic signal, but are poorly exploited by most alignment and tree building programs; (iii) even so, excluding gaps and variable regions is detrimental; (iv) disagreement among alignment programs says little about the accuracy of resulting trees.
Conclusions: This study provides the broad community relying on sequence alignment with important practical recommendations, sets superior standards for assessing alignment accuracy, and paves the way for the development of phylogenetic inference methods of significantly higher resolution.
Figures

References
-
- Blackshields G, Wallace IM, Larkin M, Higgins DG. Analysis and comparison of benchmarks for multiple sequence alignment. In Silico Biol. 2006;6:321–339. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
