

Detection of splice junctions from paired-end RNA-seq data by SpliceMap
- PMID: 20371516
- PMCID: PMC2919714
- DOI: 10.1093/nar/gkq211
Detection of splice junctions from paired-end RNA-seq data by SpliceMap
Abstract
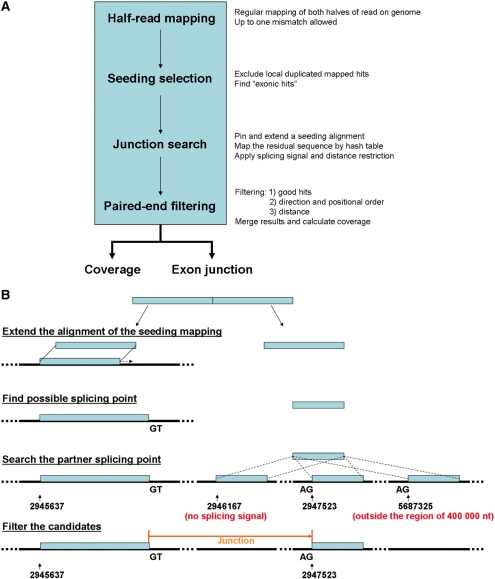
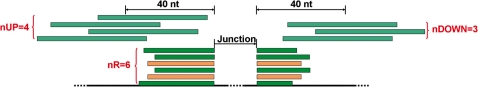
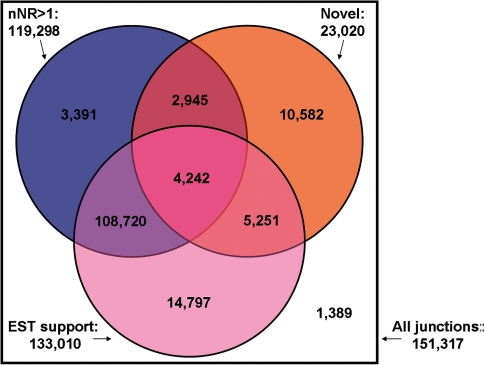
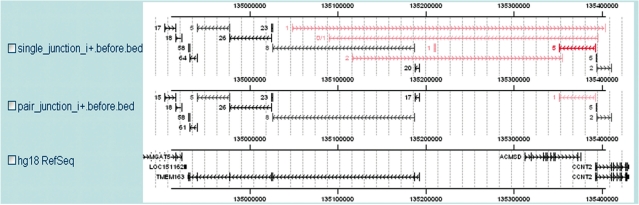
Alternative splicing is a prevalent post-transcriptional process, which is not only important to normal cellular function but is also involved in human diseases. The newly developed second generation sequencing technique provides high-throughput data (RNA-seq data) to study alternative splicing events in different types of cells. Here, we present a computational method, SpliceMap, to detect splice junctions from RNA-seq data. This method does not depend on any existing annotation of gene structures and is capable of finding novel splice junctions with high sensitivity and specificity. It can handle long reads (50-100 nt) and can exploit paired-read information to improve mapping accuracy. Several parameters are included in the output to indicate the reliability of the predicted junction and help filter out false predictions. We applied SpliceMap to analyze 23 million paired 50-nt reads from human brain tissue. The results show at this depth of sequencing, RNA-seq can support reliable detection of splice junctions except for those that are present at very low level. Compared to current methods, SpliceMap can achieve 12% higher sensitivity without sacrificing specificity.
Figures

References
-
- Matlin AJ, Clark F, Smith CW. Understanding alternative splicing: towards a cellular code. Nat. Rev. Mol. Cell. Biol. 2005;6:386–398. - PubMed
-
- Nagao K, Togawa N, Fujii K, Uchikawa H, Kohno Y, Yamada M, Miyashita T. Detecting tissue-specific alternative splicing and disease-associated aberrant splicing of the PTCH gene with exon junction microarrays. Hum. Mol. Genet. 2005;14:3379–3388. - PubMed
-
- Wang H, Hubbell E, Hu JS, Mei G, Cline M, Lu G, Clark T, Siani-Rose MA, Ares M, Kulp DC, et al. Gene structure-based splice variant deconvolution using a microarray platform. Bioinformatics. 2003;19:315–322. - PubMed
-
- Adams MD, Soares MB, Kerlavage AR, Fields C, Venter JC. Rapid cDNA sequencing (expressed sequence tags) from a directionally cloned human infant brain cDNA library. Nat. Genet. 1993;4:373–380. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
