

Estradiol-induced proliferation of papillary and follicular thyroid cancer cells is mediated by estrogen receptors alpha and beta
- PMID: 20372779
- PMCID: PMC11968770
- DOI: 10.3892/ijo_00000588
Estradiol-induced proliferation of papillary and follicular thyroid cancer cells is mediated by estrogen receptors alpha and beta
Abstract
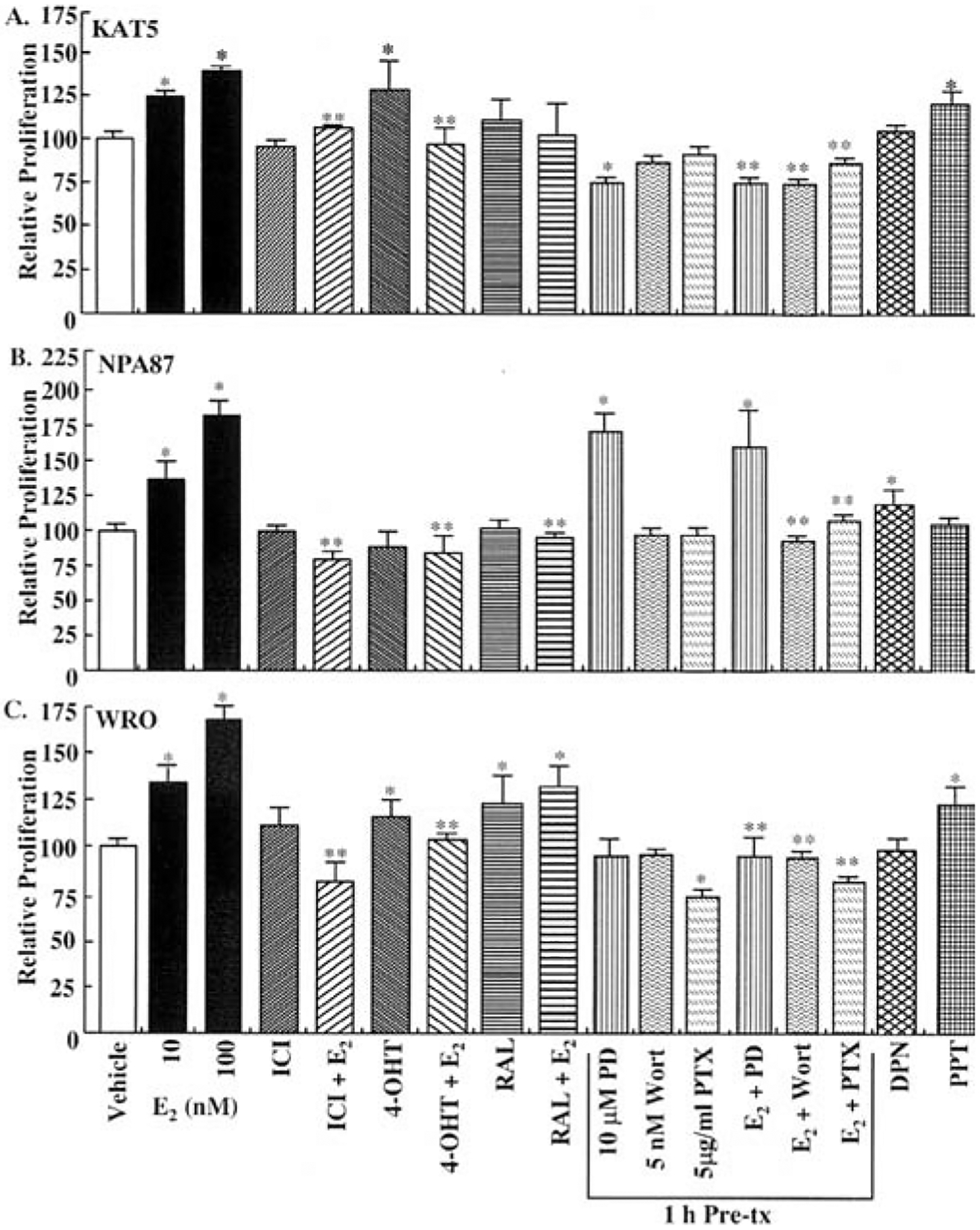
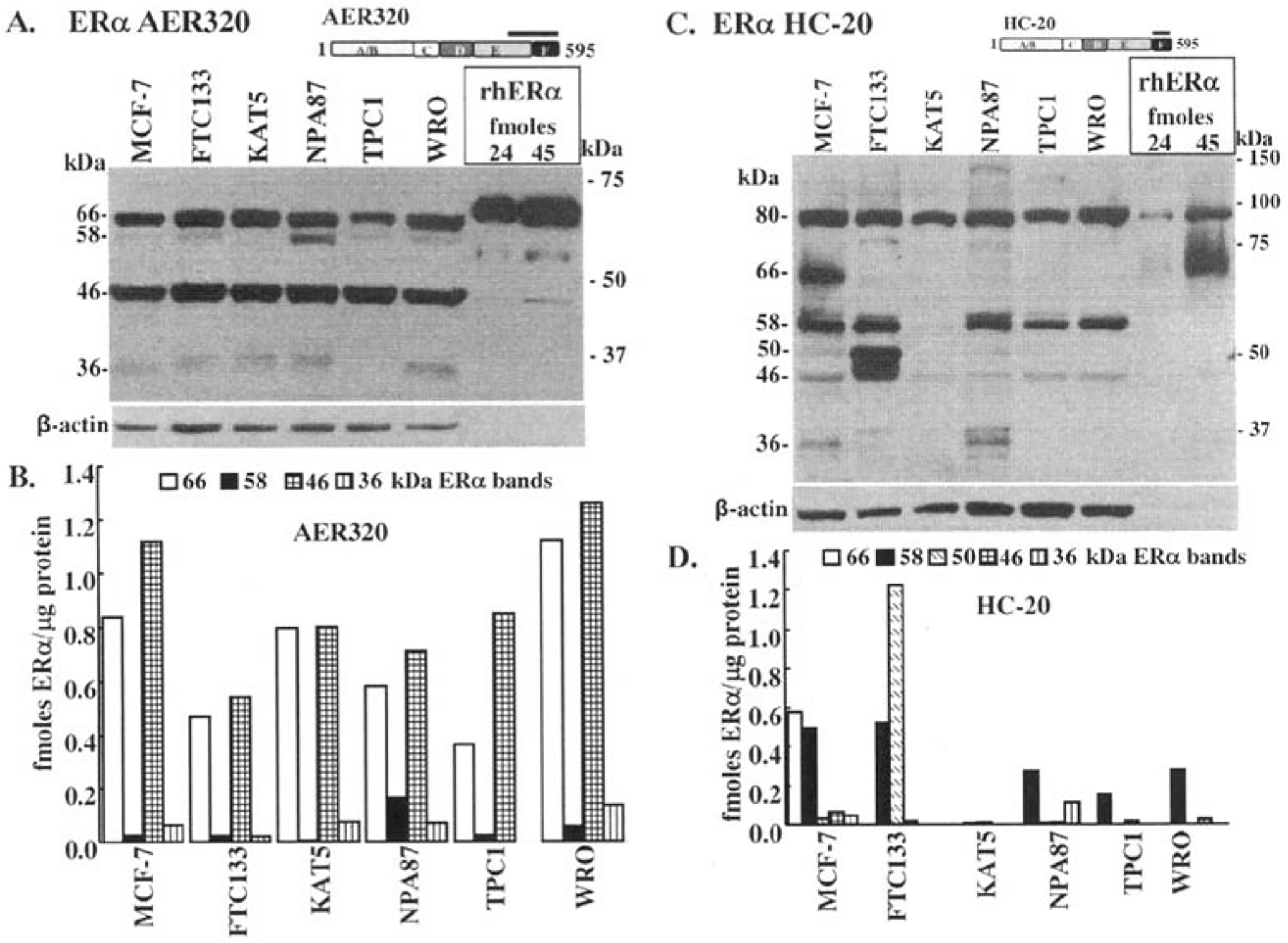
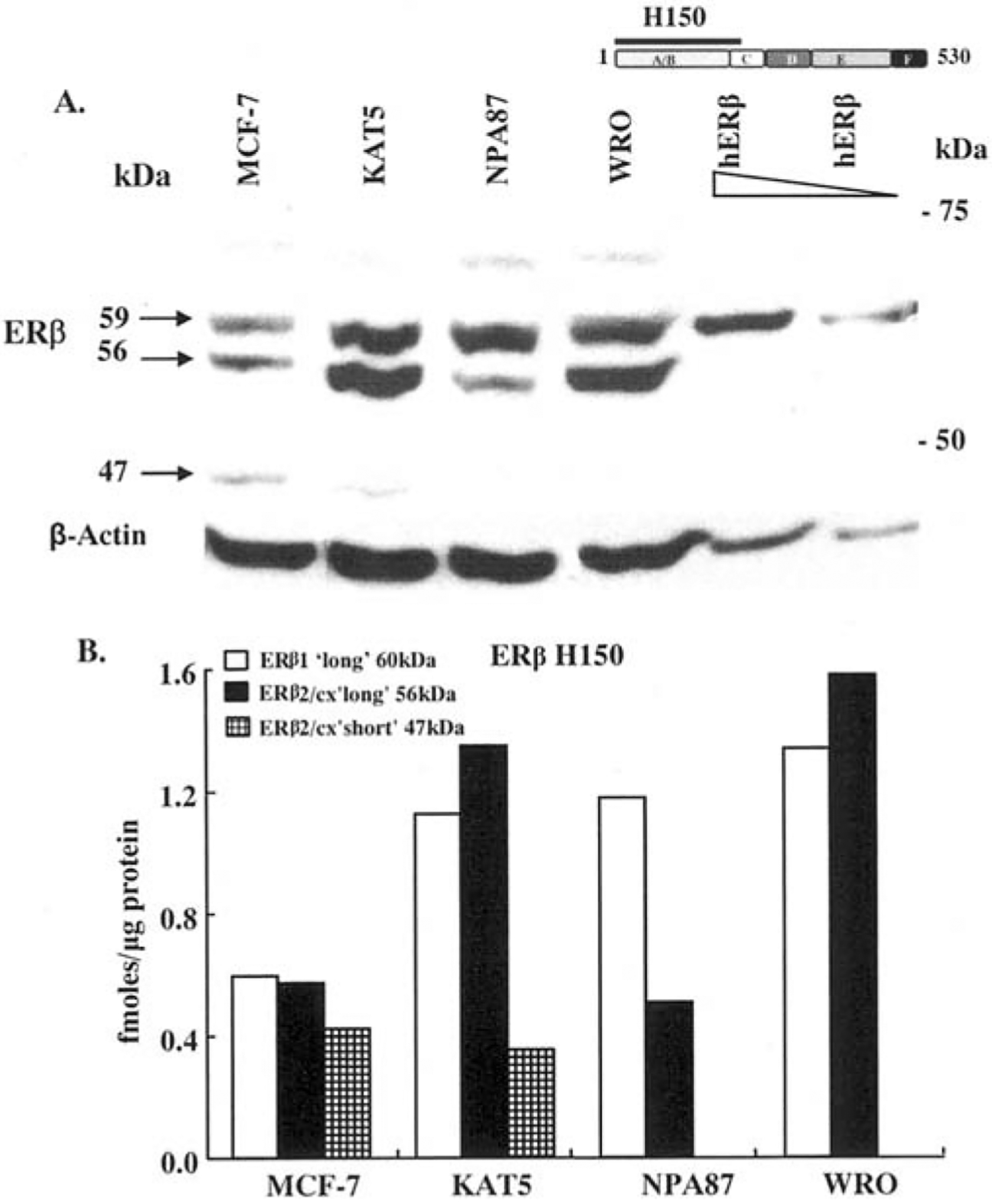
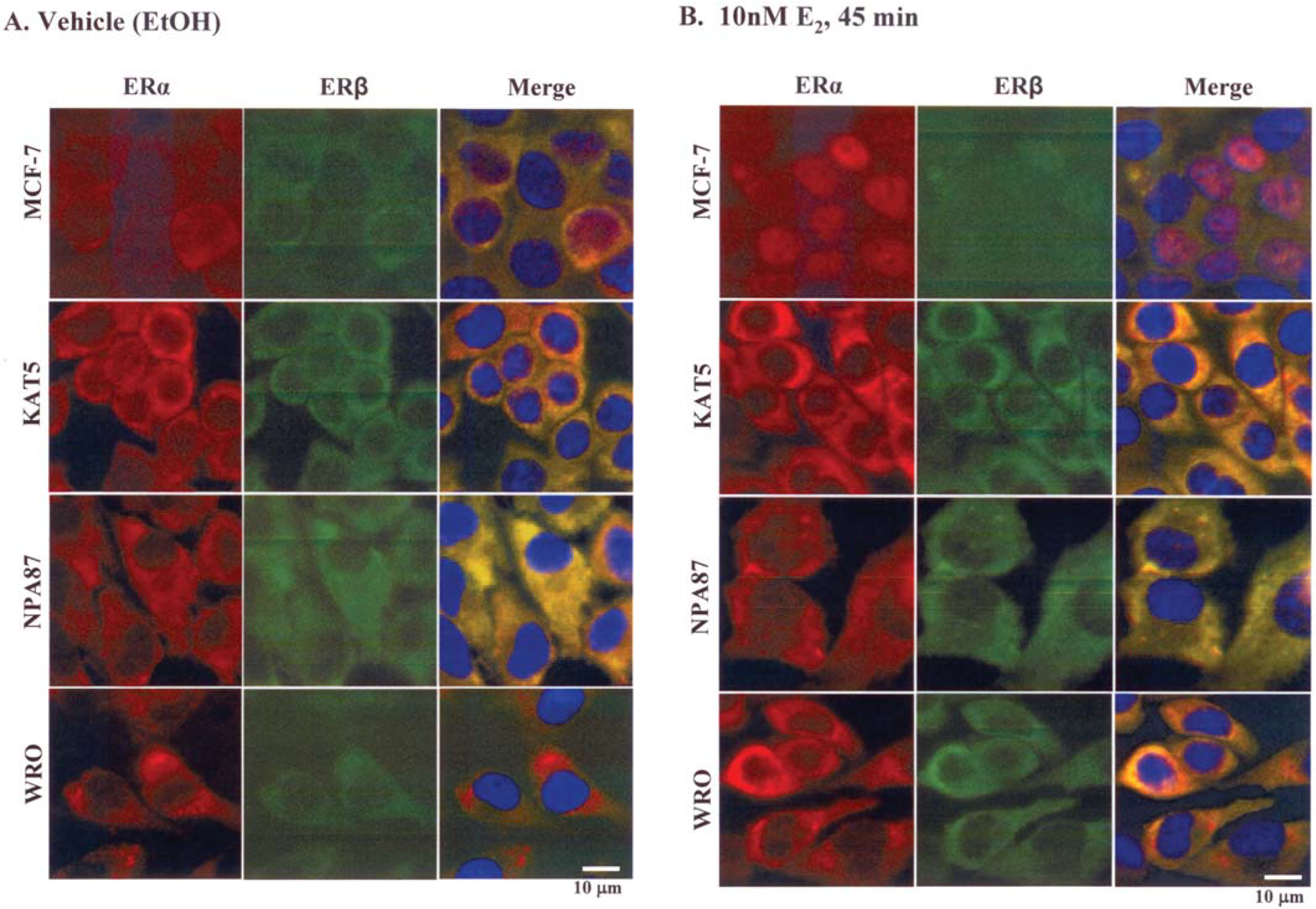
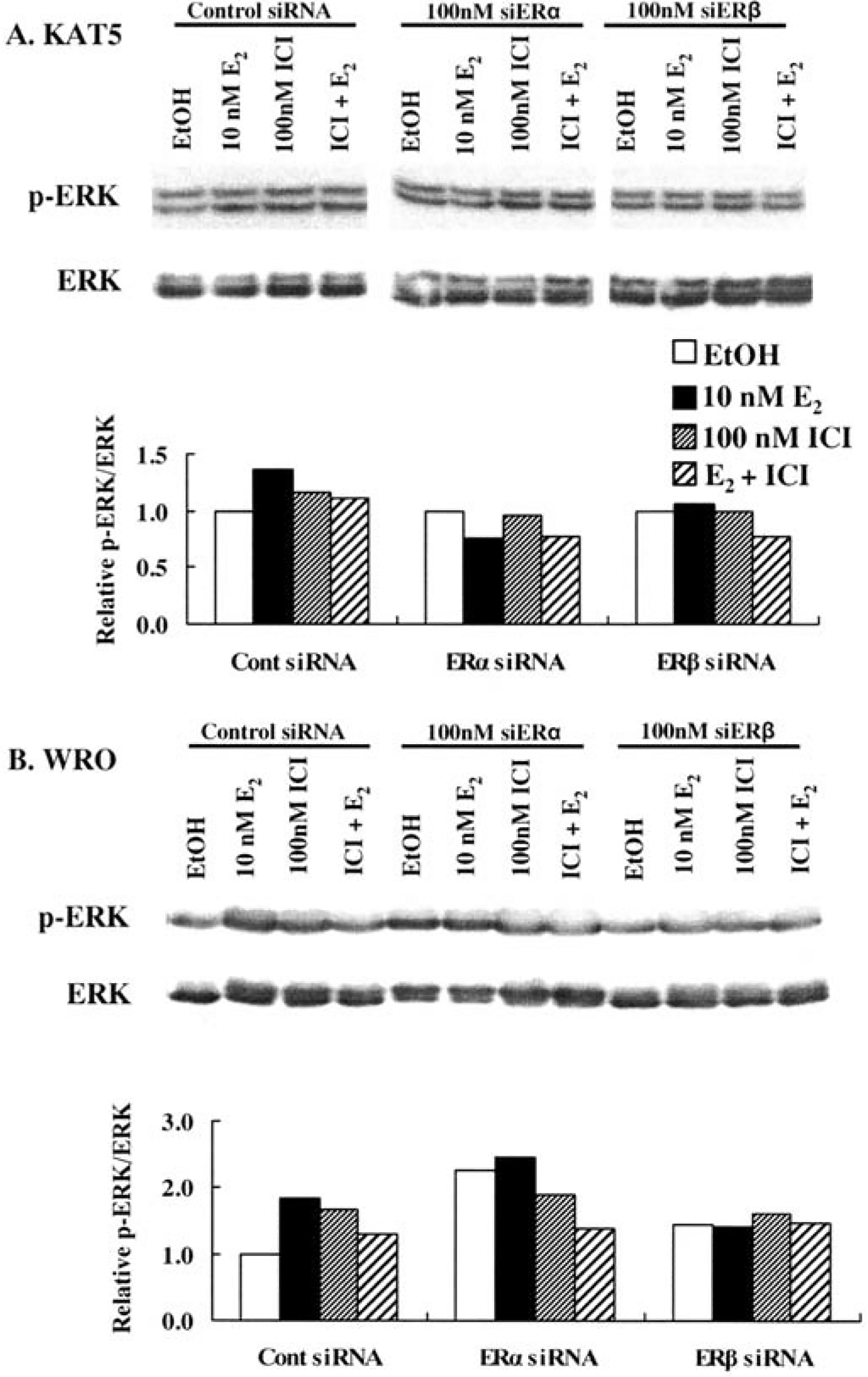
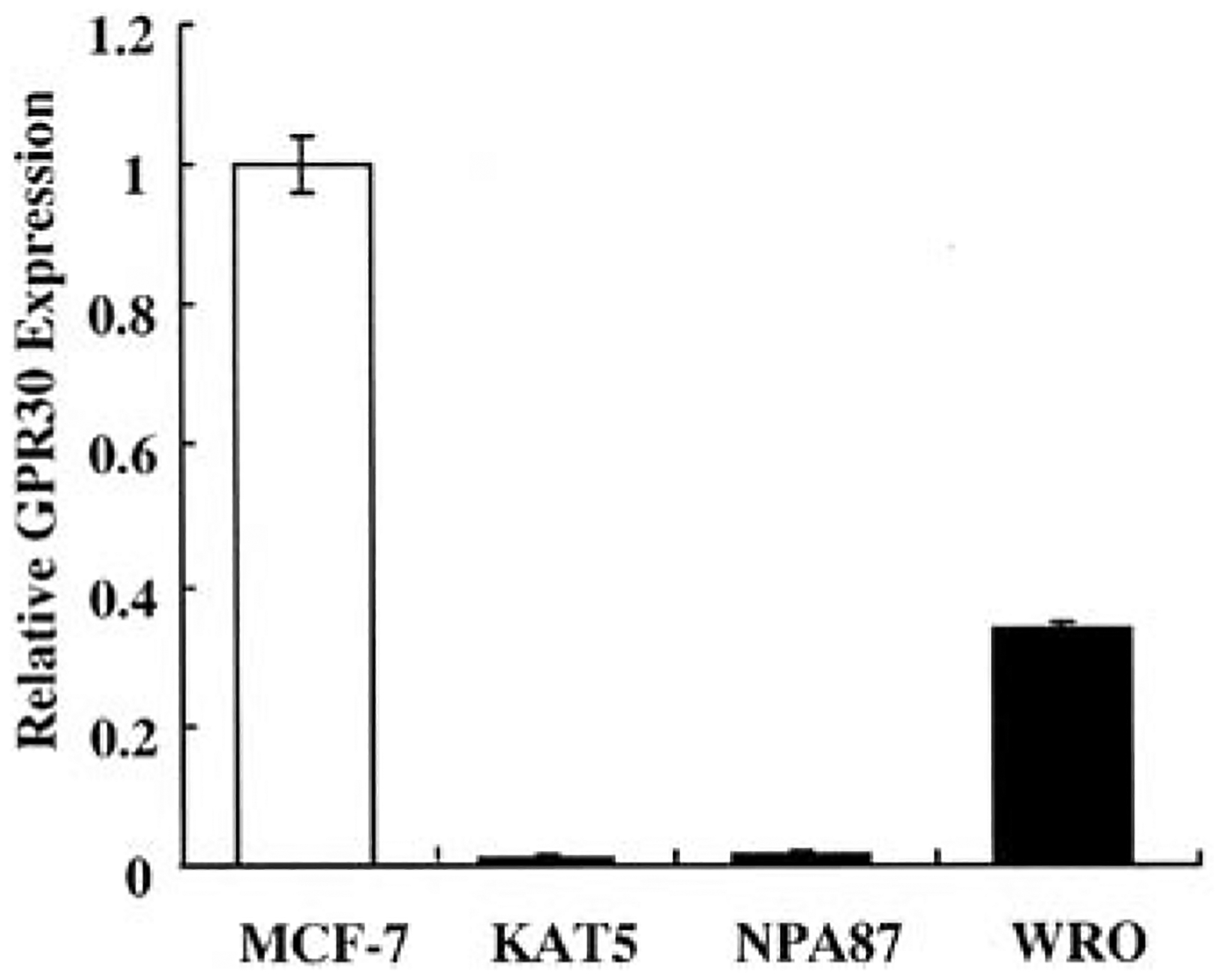
Premenopausal women are at highest risk for papillary and follicular thyroid carcinoma, implicating a role for estrogens in thyroid cancer. The expression of estrogen receptors alpha and beta (ER), the effects of estradiol (E2), selective estrogen receptor modulators (SERMs) 4-hydroxytamoxifen and raloxifene, and ER subtype selective agonists were examined in NPA87 and KAT5 papillary and WRO follicular thyroid carcinoma cell lines. All three thyroid cancer cell lines expressed full-length ERalpha and ERbeta proteins with cytoplasmic localization that was unaffected by E2. ICI 182,780 (Fulvestrant, an ER antagonist), and inhibitors of non-genomic E2-activated MAPK and PI3K signaling blocked E2-induced cell proliferation. SERMs acted in a cell line-specific manner. No E2-induced estrogen response element (ERE)-driven reporter activity was observed in transiently transfected thyroid cancer cells. However, E2 increased transcription of established endogenous E2-target genes, i.e., cathepsin D in WRO and cyclin D1 in both KAT5 and WRO cells in an ER-dependent manner as validated by inhibitor and siRNA experiments. In contrast, E2 did not increase progesterone receptor expression in the thyroid cancer cell lines. E2 stimulated phosphorylation of ERK1/2 in KAT5 and WRO cells and siERalpha or siERbeta inhibited E2-induced ERK phosphorylation. Expression of the putative membrane estrogen receptor GPR30 was detected in WRO, but not NPA87 or KAT5 cells. GPR30 expression was lower in WRO than MCF-7 human breast cancer cells. Overall, these findings suggest E2-mediated thyroid cancer cell proliferation involves ERalpha and ERbeta transcriptional and non-genomic signaling events.
Figures
References
-
- Correa P and Chen VW: Endocrine gland cancer. Cancer 75: 338–352, 1995. - PubMed
-
- Jemal A, Tiwari RC, Murray T, Ghafoor A, Samuels A, Ward E, Feuer EJ and Thun MJ: Cancer statistics, 2004. CA Cancer J Clin 54: 8–29, 2004. - PubMed
-
- Robbins J, Merino MJ, Boice JD Jr, Ron E, Ain KB, Alexander HR, Norton JA and Reynolds J: Thyroid cancer: a lethal endocrine neoplasm. Ann Intern Med 115: 133–147, 1991. - PubMed
-
- Yane K, Kitahori Y, Konishi N, Okaichi K, Ohnishi T, Miyahara H, Matsunaga T, Lin JC and Hiasa Y: Expression of the estrogen receptor in human thyroid neoplasms. Cancer Lett 84: 59–66, 1994. - PubMed
-
- Davies L and Welch HG: Increasing incidence of thyroid cancer in the United States, 1973–2002. JAMA 295: 2164–2167, 2006. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
