

Dysferlin overexpression in skeletal muscle produces a progressive myopathy
- PMID: 20373350
- PMCID: PMC3900233
- DOI: 10.1002/ana.21926
Dysferlin overexpression in skeletal muscle produces a progressive myopathy
Abstract
Objective: The dose-response effects of dysferlin transgenesis were analyzed to determine if the dysferlin-deficient myopathies are good candidates for gene replacement therapy.
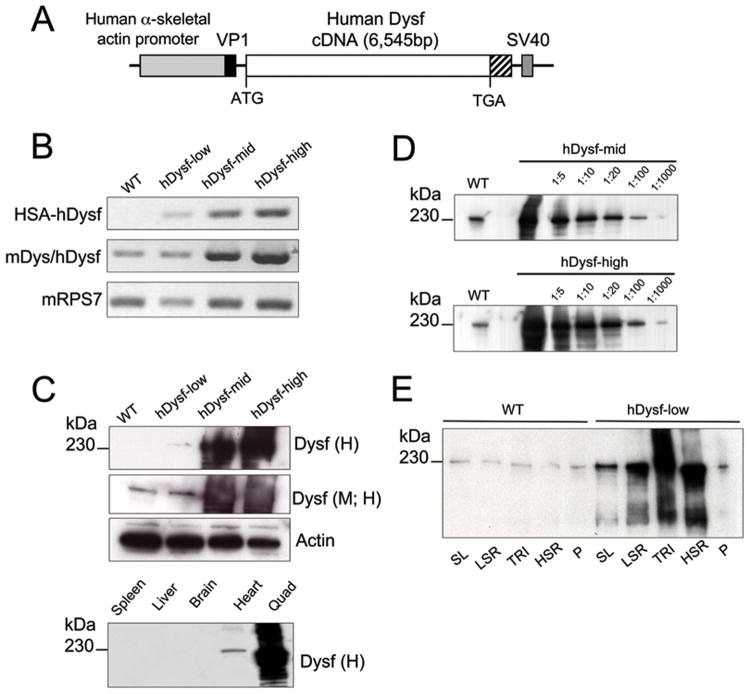
Methods: We have generated 3 lines of transgenic mice, expressing low, mid, and high levels of full-length human dysferlin from a muscle-specific promoter. Transgenic skeletal muscle was analyzed and scored for morphological and functional deficits.
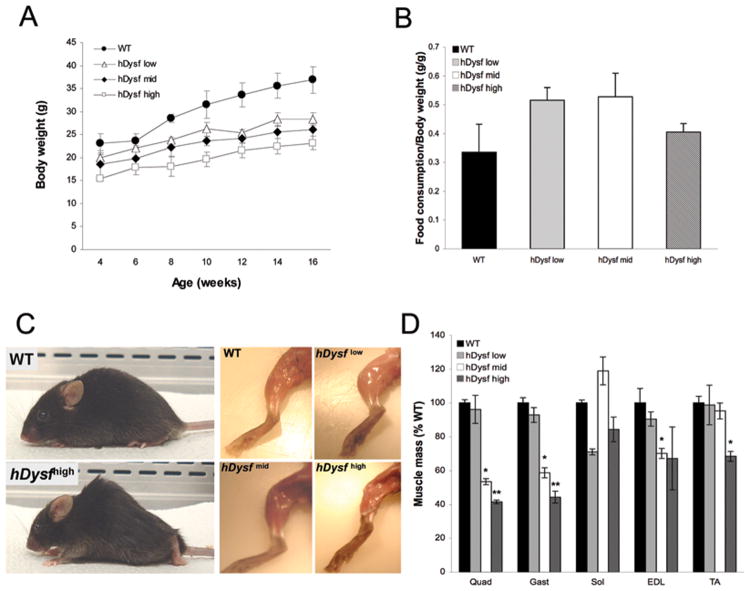
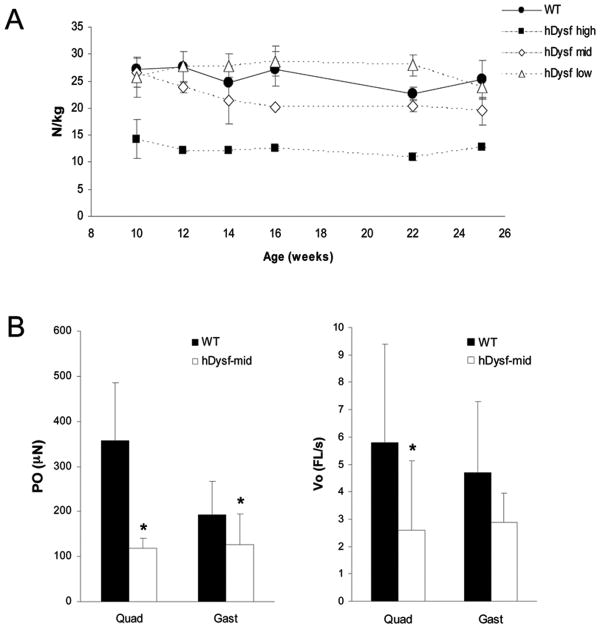
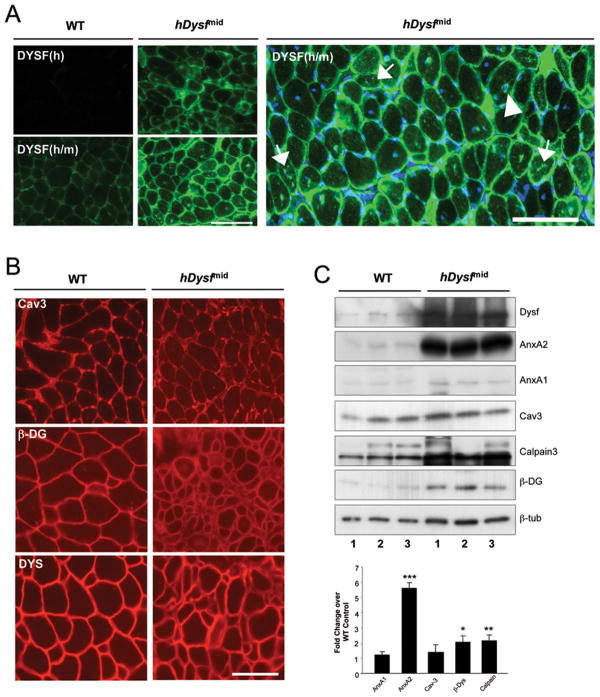
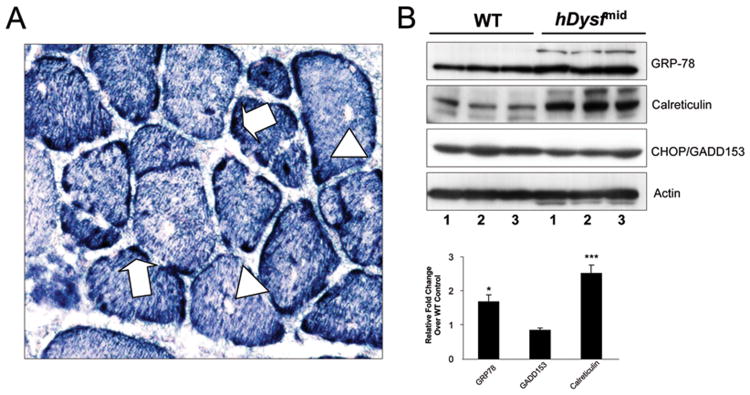
Results: Overexpression of dysferlin in mice resulted in a striking phenotype of kyphosis, irregular gait, and reduced muscle mass and strength. Moreover, protein dosage correlated with phenotype severity. In contrast to dysferlin-null skeletal muscle, no evidence of sarcolemmal impairment was revealed. Rather, increased levels of Ca(2+)-regulated, dysferlin-binding proteins and endoplasmic reticulum stress chaperone proteins were observed in muscle lysates from transgenic mice as compared with controls.
Interpretation: Expression levels of dysferlin are important for appropriate function without deleterious or cytotoxic effects. As a corollary, we propose that future endeavors in gene replacement for correction of dysferlinopathy should be tailored to take account of this.
Figures
Similar articles
-
Symptomatic dysferlin gene mutation carriers: characterization of two cases.Neurology. 2007 Apr 17;68(16):1284-9. doi: 10.1212/01.wnl.0000256768.79353.60. Epub 2007 Feb 7. Neurology. 2007. PMID: 17287450
-
A new phenotype of dysferlinopathy with congenital onset.Neuromuscul Disord. 2009 Jan;19(1):21-5. doi: 10.1016/j.nmd.2008.09.015. Epub 2008 Dec 11. Neuromuscul Disord. 2009. PMID: 19084402
-
Genetic manipulation of dysferlin expression in skeletal muscle: novel insights into muscular dystrophy.Am J Pathol. 2009 Nov;175(5):1817-23. doi: 10.2353/ajpath.2009.090107. Epub 2009 Oct 15. Am J Pathol. 2009. PMID: 19834057 Free PMC article.
-
Dysferlin function in skeletal muscle: Possible pathological mechanisms and therapeutical targets in dysferlinopathies.Exp Neurol. 2016 Sep;283(Pt A):246-54. doi: 10.1016/j.expneurol.2016.06.026. Epub 2016 Jun 25. Exp Neurol. 2016. PMID: 27349407 Review.
-
Dysferlin and the plasma membrane repair in muscular dystrophy.Trends Cell Biol. 2004 Apr;14(4):206-13. doi: 10.1016/j.tcb.2004.03.001. Trends Cell Biol. 2004. PMID: 15066638 Review.
Cited by
-
Muscle-Specific Promoters for Gene Therapy.Acta Naturae. 2021 Jan-Mar;13(1):47-58. doi: 10.32607/actanaturae.11063. Acta Naturae. 2021. PMID: 33959386 Free PMC article.
-
Continuous and low-energy 125I seed irradiation changes DNA methyltransferases expression patterns and inhibits pancreatic cancer tumor growth.J Exp Clin Cancer Res. 2011 Apr 2;30(1):35. doi: 10.1186/1756-9966-30-35. J Exp Clin Cancer Res. 2011. PMID: 21457568 Free PMC article.
-
Ozone preconditioning and exposure to ketamine attenuates hepatic inflammation in septic rats.Arch Med Sci. 2012 Nov 9;8(5):918-23. doi: 10.5114/aoms.2012.29278. Epub 2012 Jun 28. Arch Med Sci. 2012. PMID: 23185204 Free PMC article.
-
Oversized AAV transductifon is mediated via a DNA-PKcs-independent, Rad51C-dependent repair pathway.Mol Ther. 2013 Dec;21(12):2205-16. doi: 10.1038/mt.2013.184. Epub 2013 Aug 13. Mol Ther. 2013. PMID: 23939025 Free PMC article.
-
Rapid actin-cytoskeleton-dependent recruitment of plasma membrane-derived dysferlin at wounds is critical for muscle membrane repair.FASEB J. 2014 Aug;28(8):3660-70. doi: 10.1096/fj.14-250191. Epub 2014 May 1. FASEB J. 2014. PMID: 24784578 Free PMC article.
References
-
- Bashir R, Britton S, Strachan T, Keers S, Vafiadaki E, Lako M, Richard I, Marchand S, Bourg N, Argov Z, Sadeh M, Mahjneh I, Marconi G, Passos-Bueno MR, Moreira Ede S, et al. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat Genet. 1998;20(1):37–42. - PubMed
-
- Liu J, Aoki M, Illa I, Wu C, Fardeau M, Angelini C, Serrano C, Urtizberea JA, Hentati F, Hamida MB, Bohlega S, Culper EJ, Amato AA, Bossie K, Oeltjen J, et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet. 1998;20(1):31–36. - PubMed
-
- Saito A, Higuchi I, Nakagawa M, Saito M, Hirata K, Suehara M, Yoshida Y, Takahashi T, Aoki M, Osame M. Miyoshi myopathy patients with novel 5′ splicing donor site mutations showed different dysferlin immunostaining at the sarcolemma. Acta Neuropathol (Berl) 2002;104(6):615–620. - PubMed
-
- Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R, McNeil PL, Campbell KP. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423(6936):168–172. - PubMed
-
- Lennon NJ, Kho A, Bacskai BJ, Perlmutter SL, Hyman BT, Brown RH., Jr Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J Biol Chem. 2003;278(50):50466–50473. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
