

Directionality in protein fold prediction
- PMID: 20374616
- PMCID: PMC2871273
- DOI: 10.1186/1471-2105-11-172
Directionality in protein fold prediction
Abstract
Background: Ever since the ground-breaking work of Anfinsen et al. in which a denatured protein was found to refold to its native state, it has been frequently stated by the protein fold prediction community that all the information required for protein folding lies in the amino acid sequence. Recent in vitro experiments and in silico computational studies, however, have shown that cotranslation may affect the folding pathway of some proteins, especially those of ancient folds. In this paper aspects of cotranslational folding have been incorporated into a protein structure prediction algorithm by adapting the Rosetta program to fold proteins as the nascent chain elongates. This makes it possible to conduct a pairwise comparison of folding accuracy, by comparing folds created sequentially from each end of the protein.
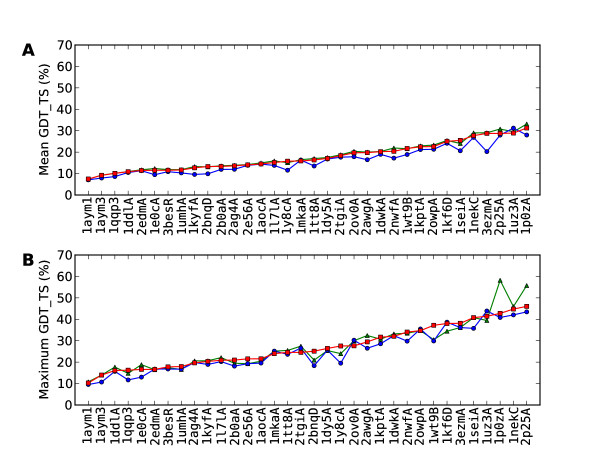
Results: A single main result emerged: in 94% of proteins analyzed, following the sense of translation, from N-terminus to C-terminus, produced better predictions than following the reverse sense of translation, from the C-terminus to N-terminus. Two secondary results emerged. First, this superiority of N-terminus to C-terminus folding was more marked for proteins showing stronger evidence of cotranslation and second, an algorithm following the sense of translation produced predictions comparable to, and occasionally better than, Rosetta.
Conclusions: There is a directionality effect in protein fold prediction. At present, prediction methods appear to be too noisy to take advantage of this effect; as techniques refine, it may be possible to draw benefit from a sequential approach to protein fold prediction.
Figures




Similar articles
-
Cotranslational protein folding--fact or fiction?Bioinformatics. 2007 Jul 1;23(13):i142-8. doi: 10.1093/bioinformatics/btm175. Bioinformatics. 2007. PMID: 17646290
-
BCL::contact-low confidence fold recognition hits boost protein contact prediction and de novo structure determination.J Comput Biol. 2010 Feb;17(2):153-68. doi: 10.1089/cmb.2009.0030. J Comput Biol. 2010. PMID: 19772383 Free PMC article.
-
Prediction of Protein Structure Using Surface Accessibility Data.Angew Chem Int Ed Engl. 2016 Sep 19;55(39):11970-4. doi: 10.1002/anie.201604788. Epub 2016 Aug 25. Angew Chem Int Ed Engl. 2016. PMID: 27560616 Free PMC article.
-
Thermodynamics of protein folding: effects of hydration and electrostatic interactions.Adv Biophys. 1994;30:105-54. doi: 10.1016/0065-227x(94)90012-4. Adv Biophys. 1994. PMID: 7709803 Review.
-
Cotranslational folding of proteins.Biochemistry (Mosc). 2000 Dec;65(12):1380-4. doi: 10.1023/a:1002800822475. Biochemistry (Mosc). 2000. PMID: 11173509 Review.
Cited by
-
Building a better fragment library for de novo protein structure prediction.PLoS One. 2015 Apr 22;10(4):e0123998. doi: 10.1371/journal.pone.0123998. eCollection 2015. PLoS One. 2015. PMID: 25901595 Free PMC article.
-
Enhancing co-translational folding of heterologous protein by deleting non-essential ribosomal proteins in Pichia pastoris.Biotechnol Biofuels. 2019 Feb 21;12:38. doi: 10.1186/s13068-019-1377-z. eCollection 2019. Biotechnol Biofuels. 2019. PMID: 30828383 Free PMC article.
-
ProtInteract: A deep learning framework for predicting protein-protein interactions.Comput Struct Biotechnol J. 2023 Jan 25;21:1324-1348. doi: 10.1016/j.csbj.2023.01.028. eCollection 2023. Comput Struct Biotechnol J. 2023. PMID: 36817951 Free PMC article.
-
Robustness by intrinsically disordered C-termini and translational readthrough.Nucleic Acids Res. 2018 Nov 2;46(19):10184-10194. doi: 10.1093/nar/gky778. Nucleic Acids Res. 2018. PMID: 30247639 Free PMC article.
-
Sequential search leads to faster, more efficient fragment-based de novo protein structure prediction.Bioinformatics. 2018 Apr 1;34(7):1132-1140. doi: 10.1093/bioinformatics/btx722. Bioinformatics. 2018. PMID: 29136098 Free PMC article.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
