

De novo motif identification improves the accuracy of predicting transcription factor binding sites in ChIP-Seq data analysis
- PMID: 20375099
- PMCID: PMC2887977
- DOI: 10.1093/nar/gkq217
De novo motif identification improves the accuracy of predicting transcription factor binding sites in ChIP-Seq data analysis
Abstract
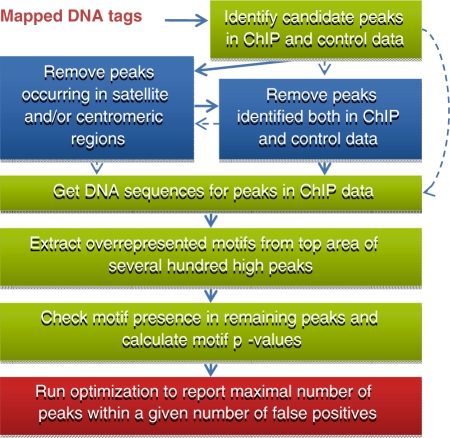
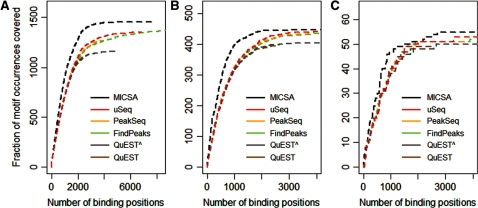
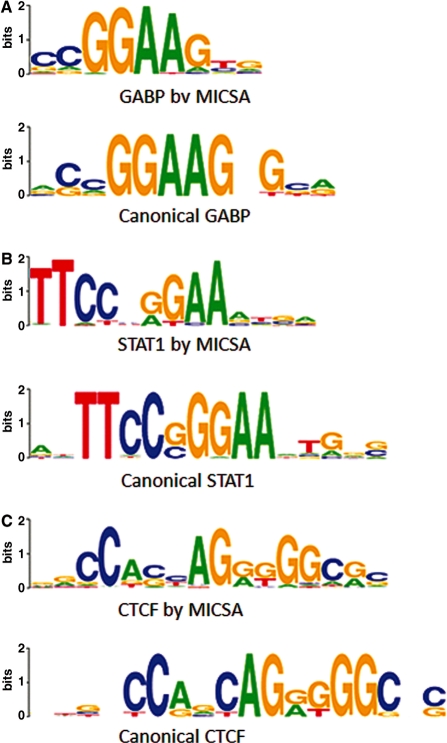
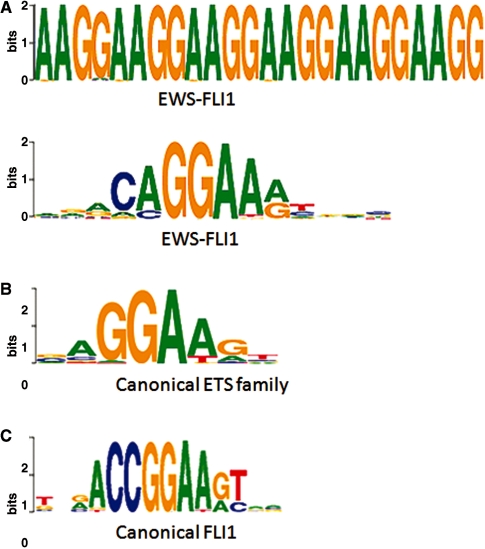
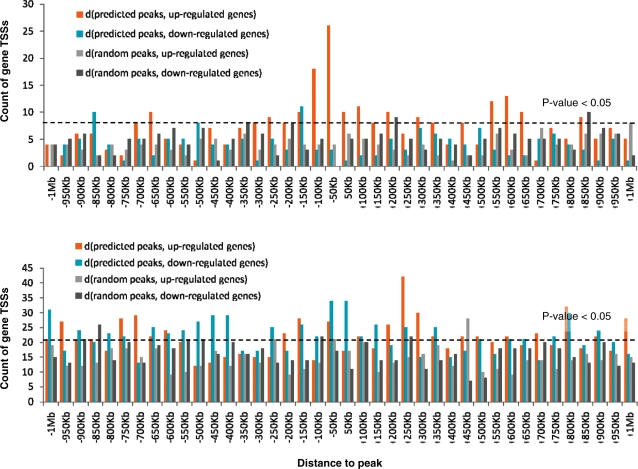
Dramatic progress in the development of next-generation sequencing technologies has enabled accurate genome-wide characterization of the binding sites of DNA-associated proteins. This technique, baptized as ChIP-Seq, uses a combination of chromatin immunoprecipitation and massively parallel DNA sequencing. Other published tools that predict binding sites from ChIP-Seq data use only positional information of mapped reads. In contrast, our algorithm MICSA (Motif Identification for ChIP-Seq Analysis) combines this source of positional information with information on motif occurrences to better predict binding sites of transcription factors (TFs). We proved the greater accuracy of MICSA with respect to several other tools by running them on datasets for the TFs NRSF, GABP, STAT1 and CTCF. We also applied MICSA on a dataset for the oncogenic TF EWS-FLI1. We discovered >2000 binding sites and two functionally different binding motifs. We observed that EWS-FLI1 can activate gene transcription when (i) its binding site is located in close proximity to the gene transcription start site (up to approximately 150 kb), and (ii) it contains a microsatellite sequence. Furthermore, we observed that sites without microsatellites can also induce regulation of gene expression--positively as often as negatively--and at much larger distances (up to approximately 1 Mb).
Figures
References
-
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. - PubMed
-
- Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. - PubMed
-
- Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods. 2007;4:651–657. - PubMed
-
- Buck MJ, Lieb JD. ChIP-chip: considerations for the design, analysis, and application of genome-wide chromatin immunoprecipitation experiments. Genomics. 2004;83:349–360. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
