

Activation of TAFI on the surface of Streptococcus pyogenes evokes inflammatory reactions by modulating the kallikrein/kinin system
- PMID: 20375563
- PMCID: PMC7312846
- DOI: 10.1159/000145543
Activation of TAFI on the surface of Streptococcus pyogenes evokes inflammatory reactions by modulating the kallikrein/kinin system
Abstract
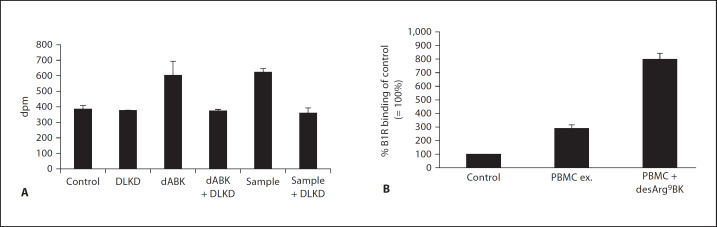
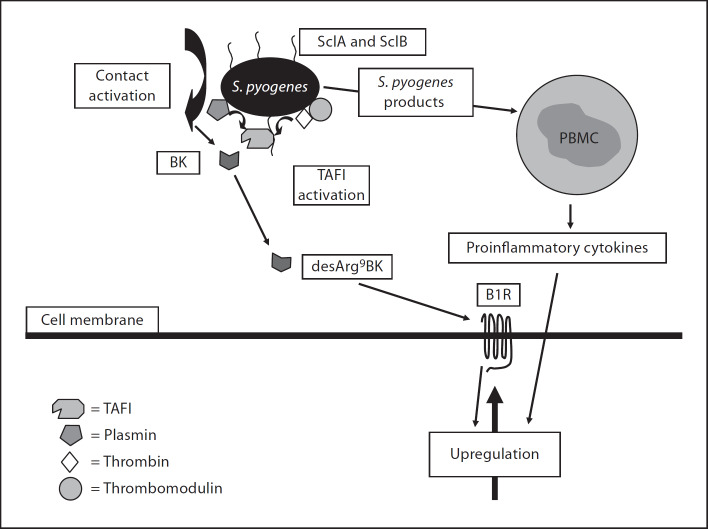
Bacteria-controlled regulation of host responses to infection is an important virulence mechanism that has been demonstrated to contribute to disease progression. Here we report that the human pathogen Streptococcus pyogenes employs the procarboxypeptidase TAFI (thrombin-activatable fibrinolysis inhibitor) to modulate the kallikrein/kinin system. To this end, bacteria initiate a chain of events starting with the recruitment and activation of TAFI. This is followed by the assembly and induction of the contact system at the streptococcal surface, eventually triggering the release of bradykinin (BK). BK is then carboxyterminally truncated by activated TAFI, which converts the peptide from a kinin B(2) receptor ligand to a kinin B(1) receptor (B1R) agonist. Finally, we show that streptococcal supernatants indirectly amplify the B1R response as they act on peripheral blood mononuclear cells to secrete inflammatory cytokines that in turn stimulate upregulation of the B1R on human fibroblasts. Taken together our findings implicate a critical and novel role for streptococci-bound TAFI, as it processes BK to a B1R agonist at the bacterial surface and thereby may redirect inflammation from a transient to a chronic state.
Copyright 2008 S. Karger AG, Basel.
Figures
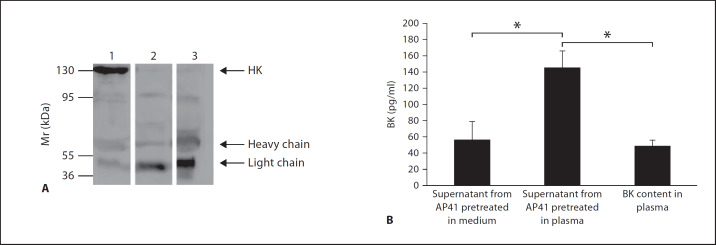
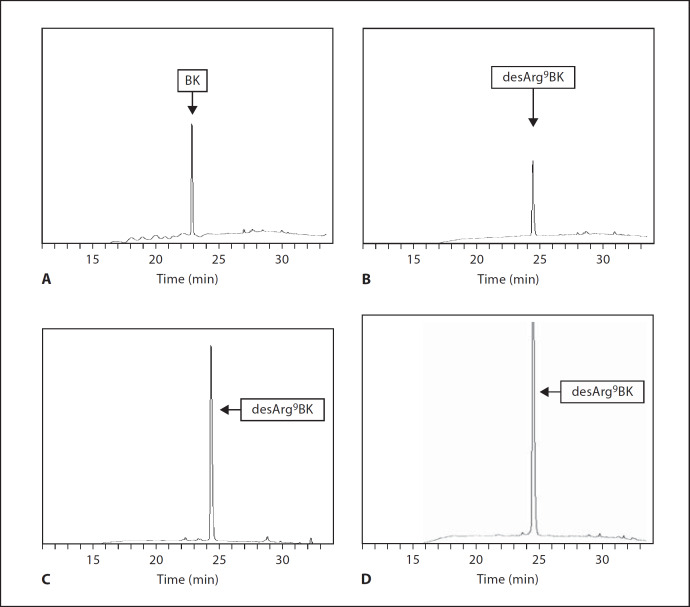

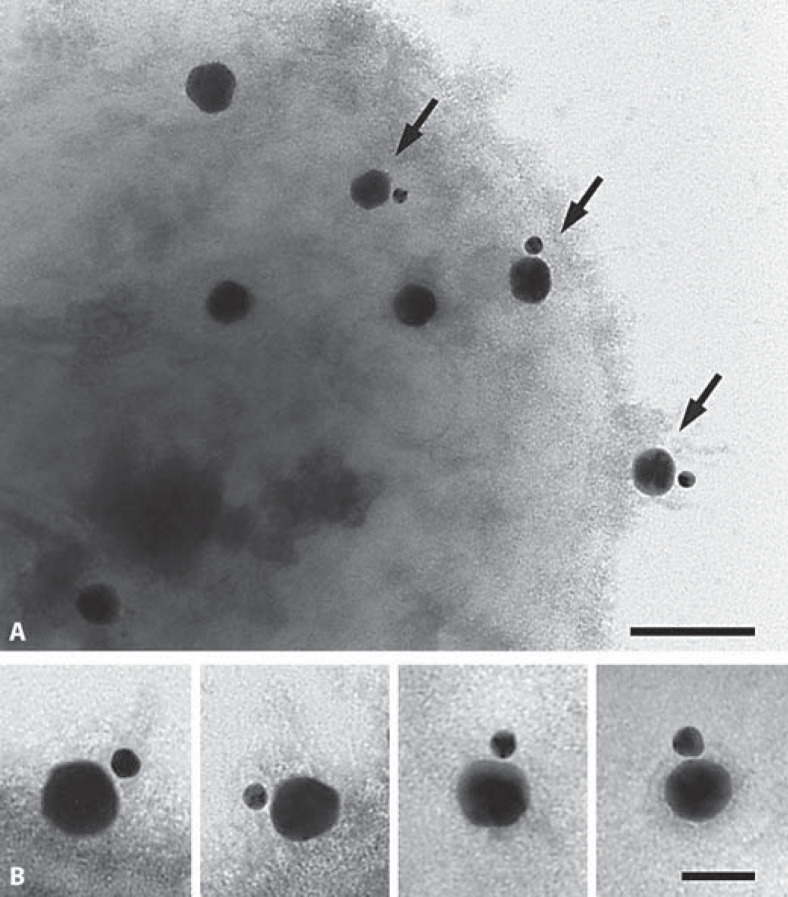
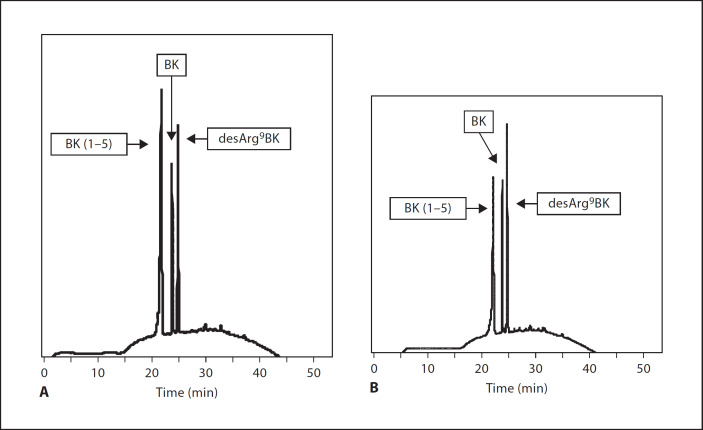
Similar articles
-
Understanding the Pathophysiology of COVID-19: Could the Contact System Be the Key?Front Immunol. 2020 Aug 11;11:2014. doi: 10.3389/fimmu.2020.02014. eCollection 2020. Front Immunol. 2020. PMID: 32849666 Free PMC article.
-
The kallikrein-Kinin system modulates the progression of colorectal liver metastases in a mouse model.BMC Cancer. 2018 Apr 4;18(1):382. doi: 10.1186/s12885-018-4260-6. BMC Cancer. 2018. PMID: 29618333 Free PMC article.
-
Bradykinin receptor in immune-mediated renal tubular injury in trichloroethylene-sensitized mice: Impact on NF-κB signaling pathway.J Immunotoxicol. 2018 Dec;15(1):126-136. doi: 10.1080/1547691X.2018.1532974. J Immunotoxicol. 2018. PMID: 30409067
-
[A role of kallikrein-kinin system in gut diseases].Wiad Lek. 2005;58(5-6):331-4. Wiad Lek. 2005. PMID: 16238127 Review. Polish.
-
G Protein-Coupled Kinin Receptors and Immunity Against Pathogens.Adv Immunol. 2017;136:29-84. doi: 10.1016/bs.ai.2017.05.007. Epub 2017 Aug 23. Adv Immunol. 2017. PMID: 28950949 Review.
Cited by
-
Enhanced activity of transforming growth factor β1 (TGF-β1) bound to cartilage oligomeric matrix protein.J Biol Chem. 2011 Dec 16;286(50):43250-8. doi: 10.1074/jbc.M111.234716. Epub 2011 Sep 22. J Biol Chem. 2011. PMID: 21940632 Free PMC article.
-
Immunohaemostasis: a new view on haemostasis during sepsis.Ann Intensive Care. 2017 Dec 2;7(1):117. doi: 10.1186/s13613-017-0339-5. Ann Intensive Care. 2017. PMID: 29197958 Free PMC article. Review.
-
Binding of albumin promotes bacterial survival at the epithelial surface.J Biol Chem. 2011 Jan 28;286(4):2469-76. doi: 10.1074/jbc.M110.148171. Epub 2010 Nov 22. J Biol Chem. 2011. PMID: 21098039 Free PMC article.
-
Midkine and pleiotrophin have bactericidal properties: preserved antibacterial activity in a family of heparin-binding growth factors during evolution.J Biol Chem. 2010 May 21;285(21):16105-15. doi: 10.1074/jbc.M109.081232. Epub 2010 Mar 22. J Biol Chem. 2010. PMID: 20308059 Free PMC article.
-
Corruption of innate immunity by bacterial proteases.J Innate Immun. 2009;1(2):70-87. doi: 10.1159/000181144. J Innate Immun. 2009. PMID: 19756242 Free PMC article. Review.
References
-
- Sriskandan S, Altmann D. The immunology of sepsis. J Pathol. 2008;214:211–223. - PubMed
-
- Carapetis JR, Steer AC, Mulholland EK, Weber M. The global burden of group A streptococcal diseases. Lancet Infect Dis. 2005;5:685–694. - PubMed
-
- Sriskandan S, Faulkner L, Hopkins P. Streptococcus pyogenes: insight into the function of the streptococcal superantigens. Int J Biochem Cell Biol. 2007;39:12–19. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
