

Recruitment of MBD1 to target genes requires sequence-specific interaction of the MBD domain with methylated DNA
- PMID: 20378711
- PMCID: PMC2919722
- DOI: 10.1093/nar/gkq228
Recruitment of MBD1 to target genes requires sequence-specific interaction of the MBD domain with methylated DNA
Abstract
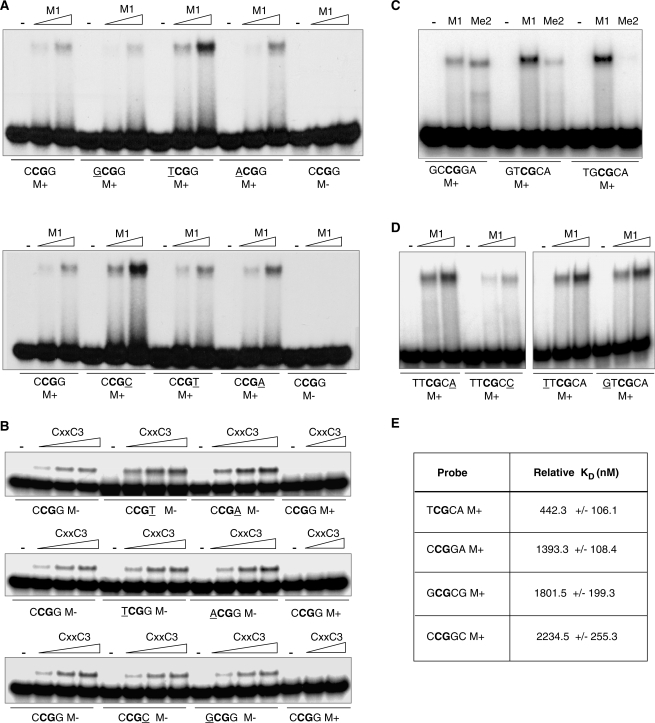
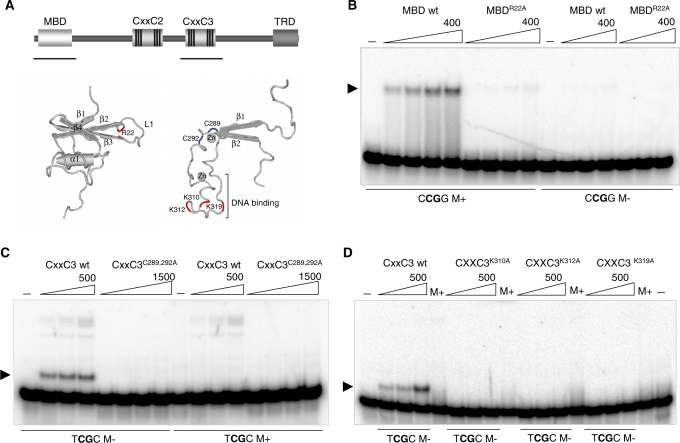
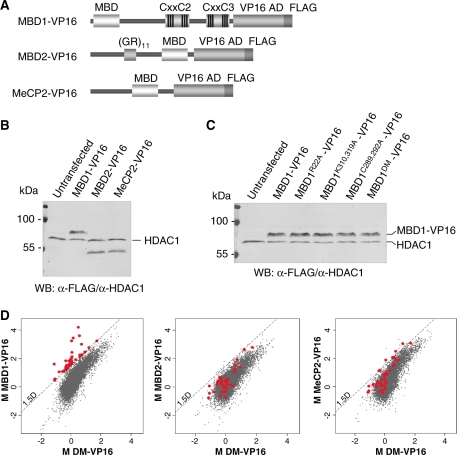
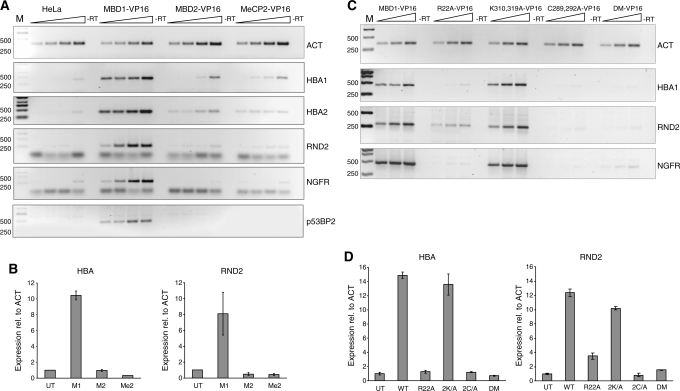
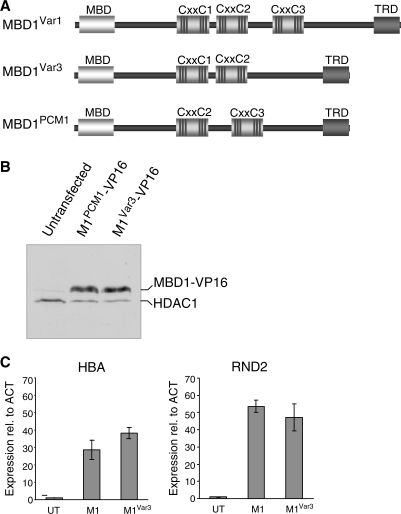
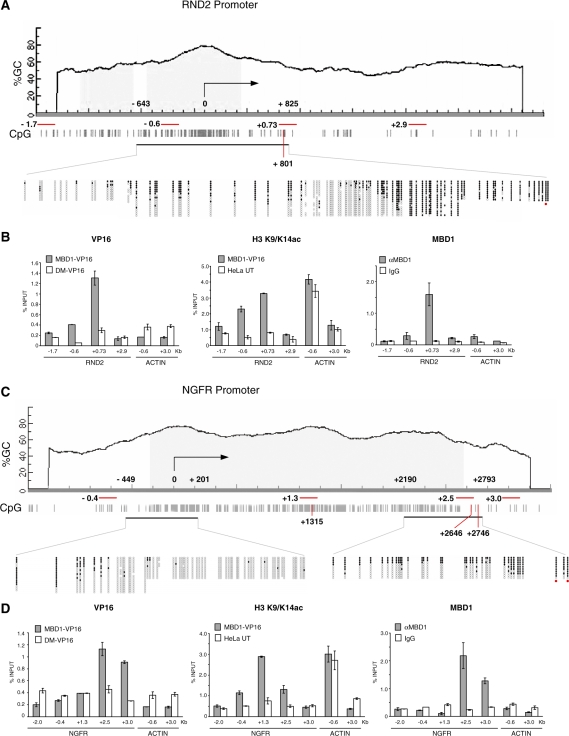
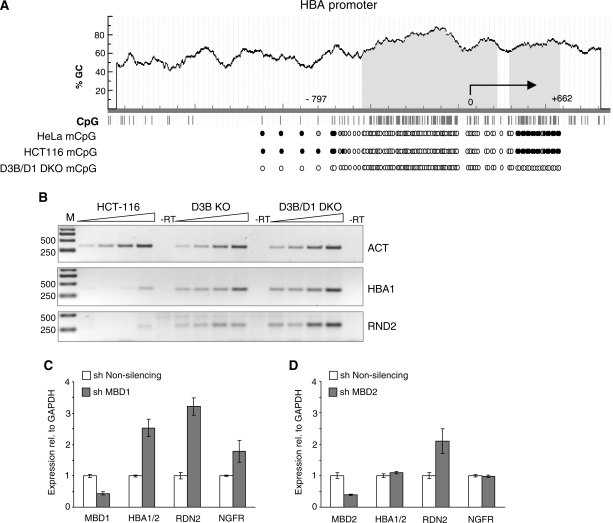
MBD1, a member of the methyl-CpG-binding domain family of proteins, has been reported to repress transcription of methylated and unmethylated promoters. As some MBD1 isoforms contain two DNA-binding domains-an MBD, which recognizes methylated DNA; and a CXXC3 zinc finger, which binds unmethylated CpG-it is unclear whether these two domains function independently of each other or if they cooperate in facilitating recruitment of MBD1 to particular genomic loci. In this report we investigate DNA-binding specificity of MBD and CXXC3 domains in vitro and in vivo. We find that the methyl-CpG-binding domain of MBD1 binds more efficiently to methylated DNA within a specific sequence context. We identify genes that are targeted by MBD1 in human cells and demonstrate that a functional MBD domain is necessary and sufficient for recruitment of MBD1 to specific sites at these loci, while DNA binding by the CXXC3 motif is largely dispensable. In summary, the binding preferences of MBD1, although dependent upon the presence of methylated DNA, are clearly distinct from those of other methyl-CpG-binding proteins, MBD2 and MeCP2.
Figures
Similar articles
-
Mechanism of transcriptional regulation by methyl-CpG binding protein MBD1.Mol Cell Biol. 2000 Jul;20(14):5107-18. doi: 10.1128/MCB.20.14.5107-5118.2000. Mol Cell Biol. 2000. PMID: 10866667 Free PMC article.
-
Regulation of transcription and chromatin by methyl-CpG binding protein MBD1.Brain Dev. 2001 Dec;23 Suppl 1:S174-6. doi: 10.1016/s0387-7604(01)00348-5. Brain Dev. 2001. PMID: 11738867 Review.
-
Methylation-mediated transcriptional silencing in euchromatin by methyl-CpG binding protein MBD1 isoforms.Mol Cell Biol. 1999 Sep;19(9):6415-26. doi: 10.1128/MCB.19.9.6415. Mol Cell Biol. 1999. PMID: 10454587 Free PMC article.
-
Methyl-CpG binding domain proteins and their involvement in the regulation of the MAGE-A1, MAGE-A2, MAGE-A3, and MAGE-A12 gene promoters.Mol Cancer Res. 2007 Jul;5(7):749-59. doi: 10.1158/1541-7786.MCR-06-0364. Mol Cancer Res. 2007. PMID: 17634428
-
Methyl-CpG-binding domain proteins: readers of the epigenome.Epigenomics. 2015;7(6):1051-73. doi: 10.2217/epi.15.39. Epub 2015 Apr 30. Epigenomics. 2015. PMID: 25927341 Review.
Cited by
-
Introduction--Epiphanies in epigenetics.Prog Mol Biol Transl Sci. 2011;101:1-21. doi: 10.1016/B978-0-12-387685-0.00001-9. Prog Mol Biol Transl Sci. 2011. PMID: 21507348 Free PMC article.
-
Recognition of methylated DNA through methyl-CpG binding domain proteins.Nucleic Acids Res. 2012 Mar;40(6):2747-58. doi: 10.1093/nar/gkr1057. Epub 2011 Nov 22. Nucleic Acids Res. 2012. PMID: 22110028 Free PMC article.
-
A/T Run Geometry of B-form DNA Is Independent of Bound Methyl-CpG Binding Domain, Cytosine Methylation and Flanking Sequence.Sci Rep. 2016 Aug 9;6:31210. doi: 10.1038/srep31210. Sci Rep. 2016. PMID: 27502833 Free PMC article.
-
DNA methylation regulates discrimination of enhancers from promoters through a H3K4me1-H3K4me3 seesaw mechanism.BMC Genomics. 2017 Dec 12;18(1):964. doi: 10.1186/s12864-017-4353-7. BMC Genomics. 2017. PMID: 29233090 Free PMC article.
-
On how mammalian transcription factors recognize methylated DNA.Epigenetics. 2013 Feb;8(2):131-7. doi: 10.4161/epi.23632. Epub 2013 Jan 16. Epigenetics. 2013. PMID: 23324617 Free PMC article. Review.
References
-
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. - PubMed
-
- Chan SW, Henderson IR, Jacobsen SE. Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat. Rev. Genet. 2005;6:351–360. - PubMed
-
- Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005;74:481–514. - PubMed
-
- Costello JF, Fruhwald MC, Smiraglia DJ, Rush LJ, Robertson GP, Gao X, Wright FA, Feramisco JD, Peltomaki P, Lang JC, et al. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat. Genet. 2000;24:132–138. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
