

Adhesion to the extracellular matrix is required for interleukin-1 beta actions leading to reactive phenotype in rat astrocytes
- PMID: 20380881
- PMCID: PMC3507629
- DOI: 10.1016/j.mcn.2010.03.013
Adhesion to the extracellular matrix is required for interleukin-1 beta actions leading to reactive phenotype in rat astrocytes
Abstract
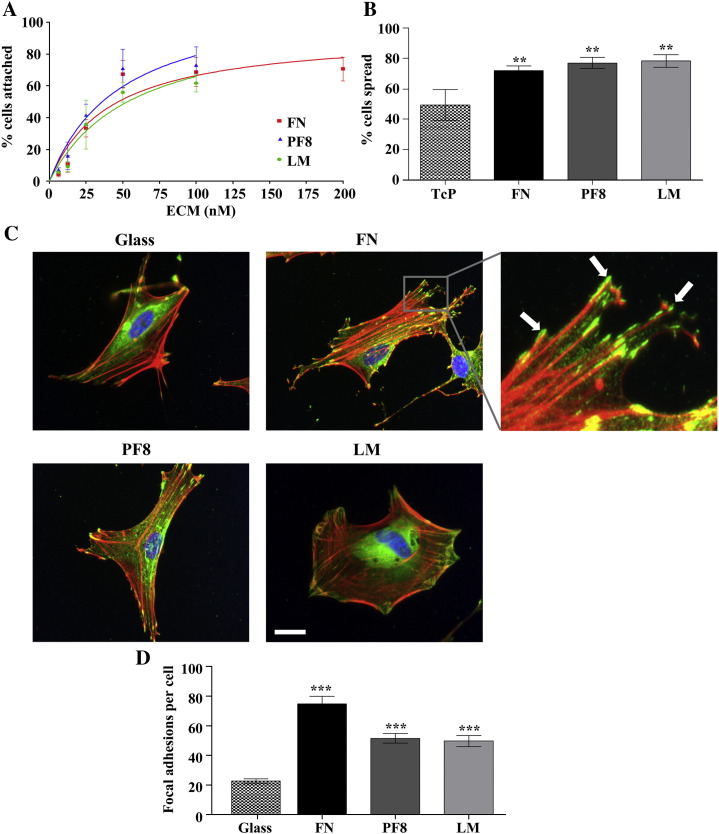
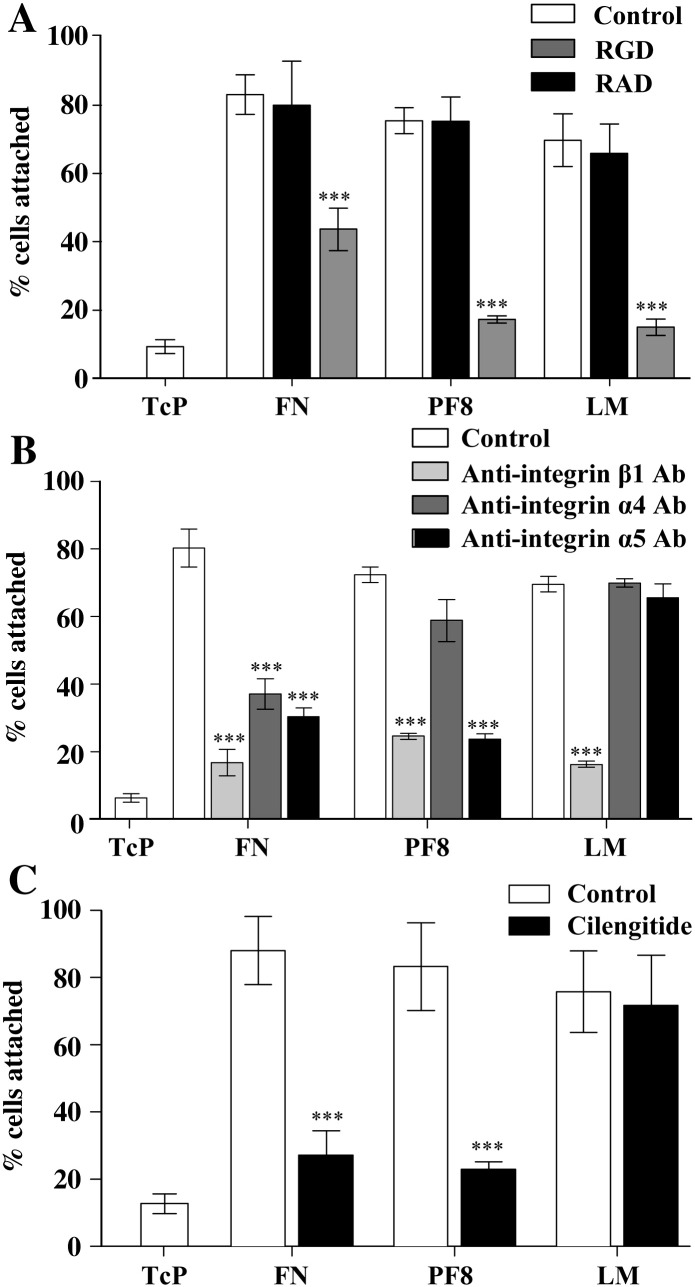
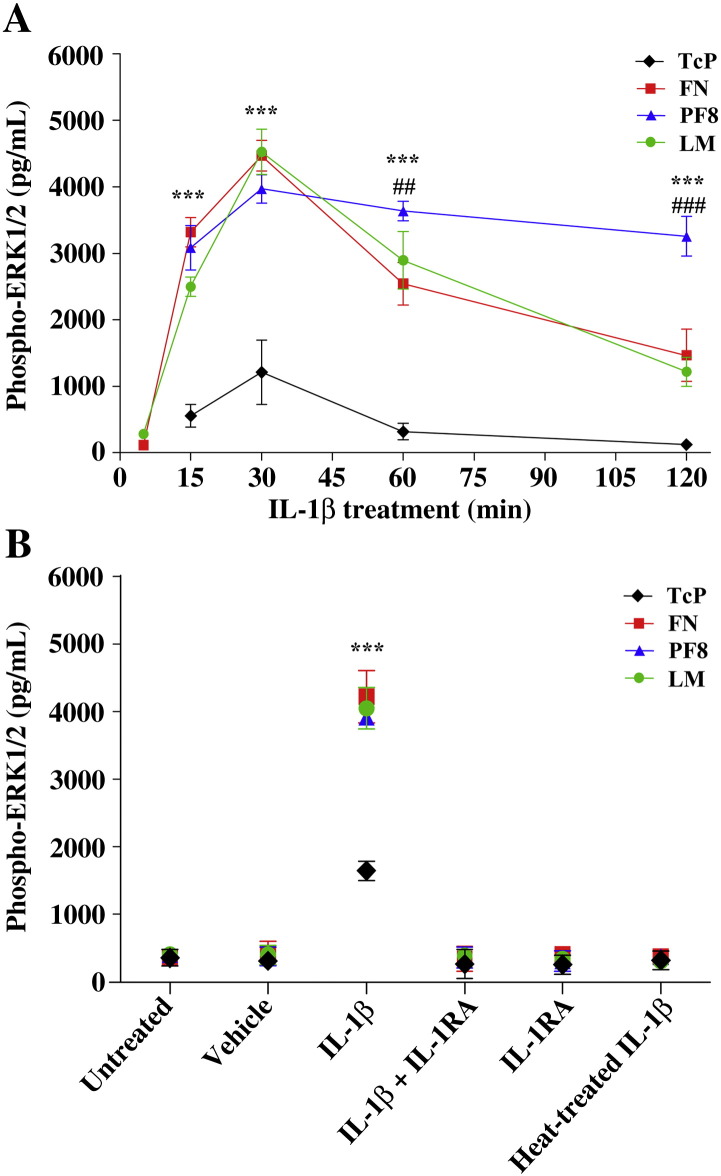
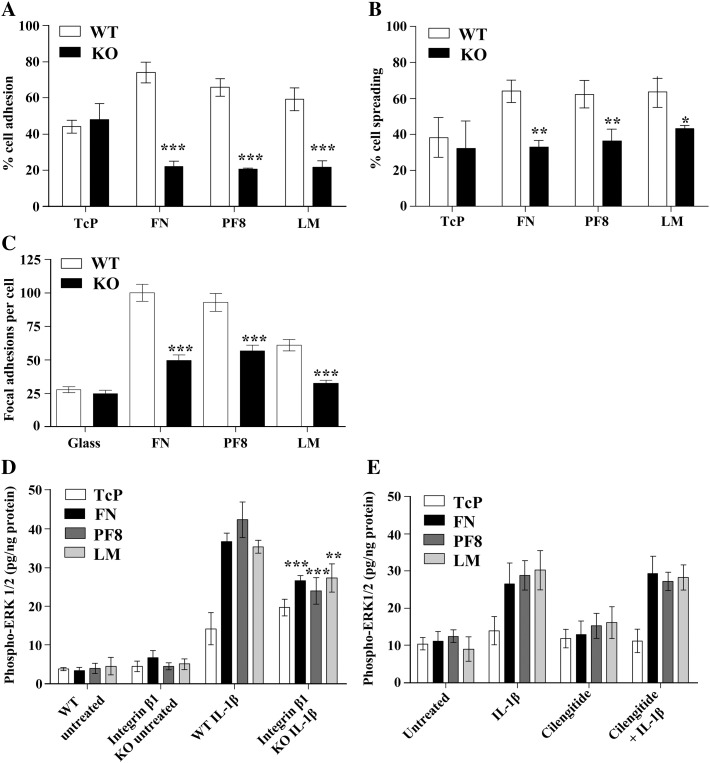
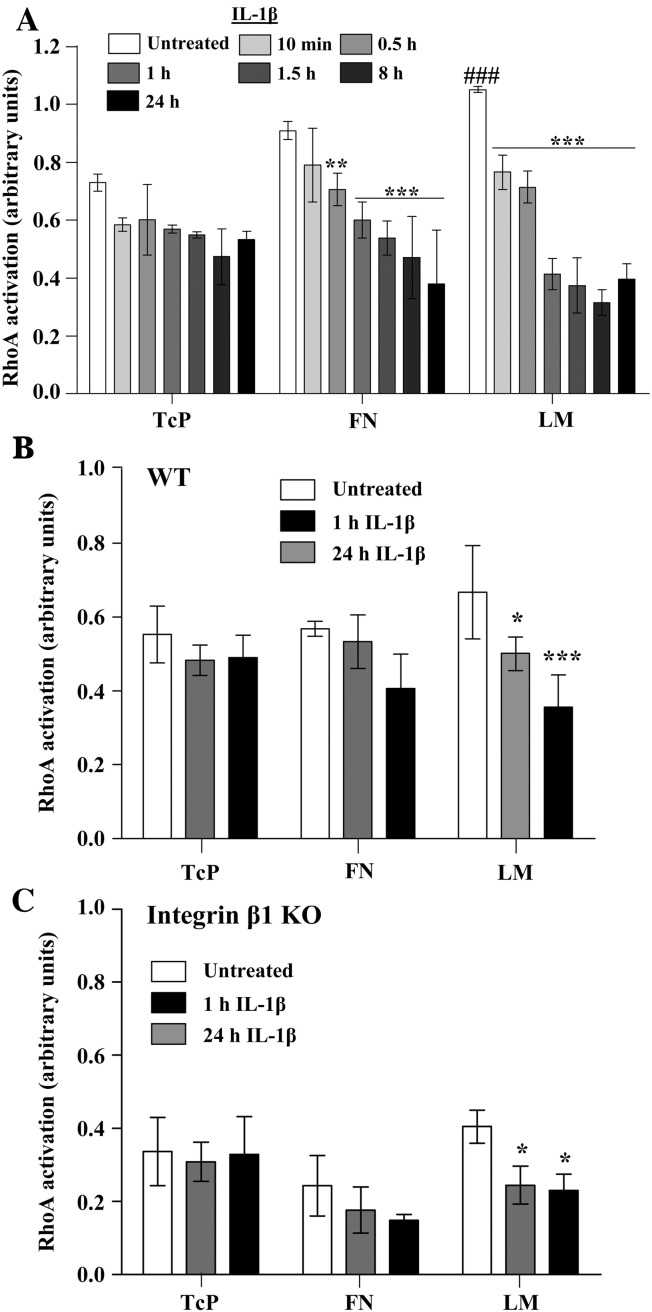
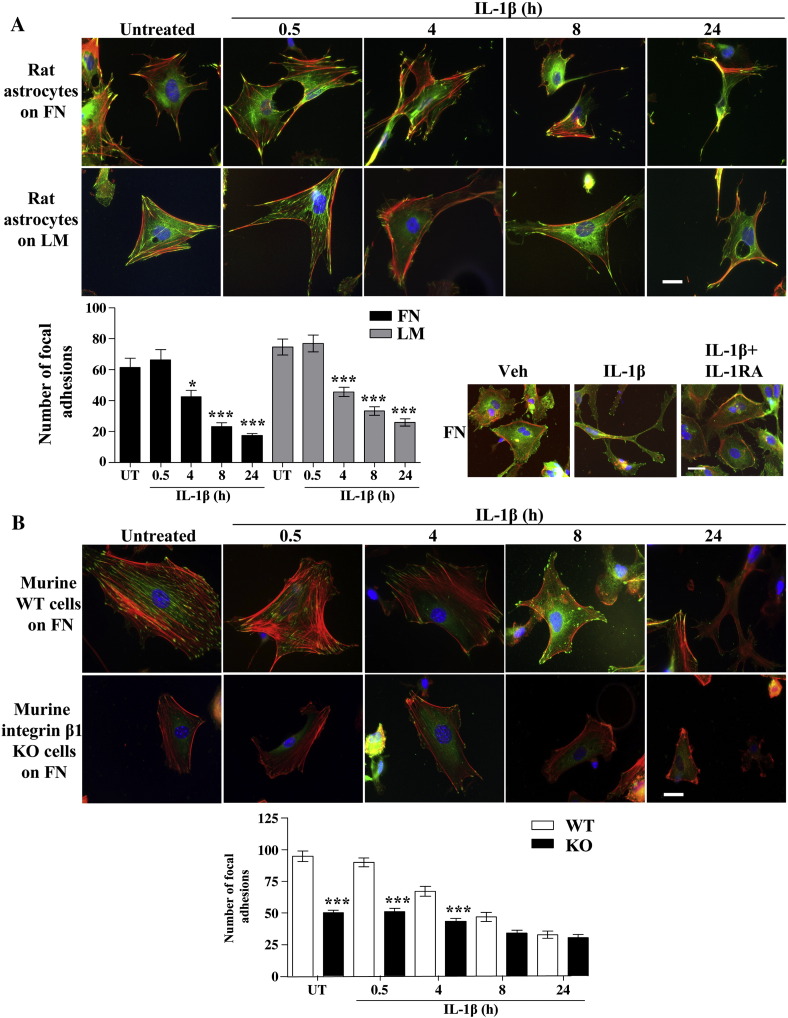
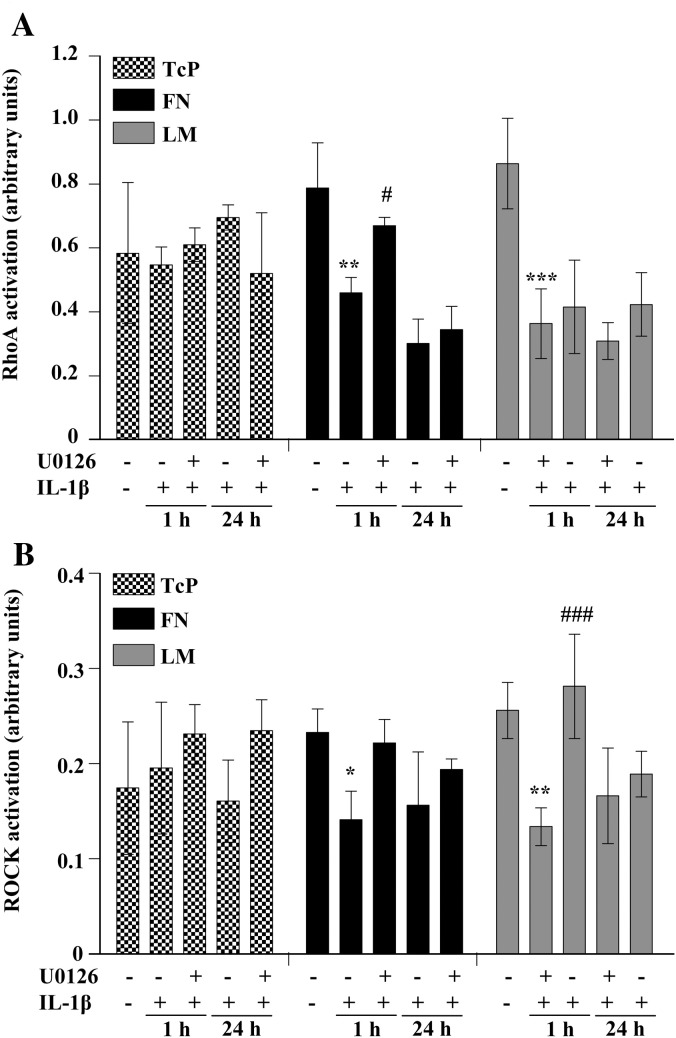
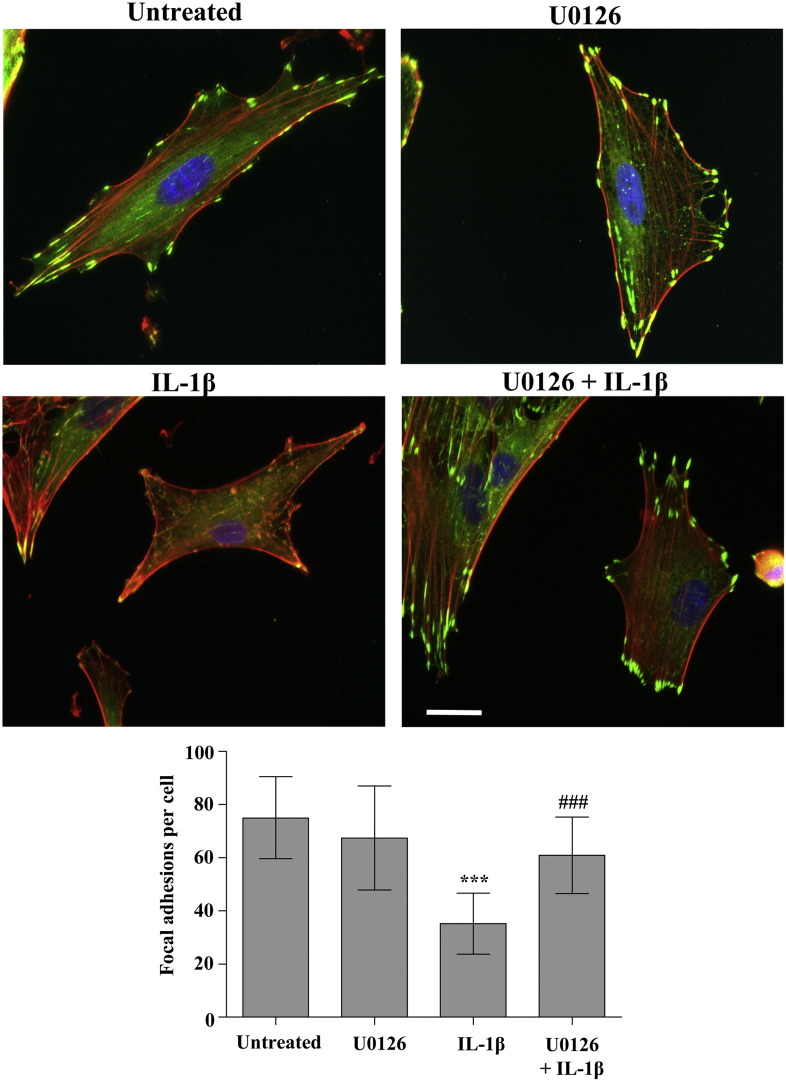
The extracellular matrix (ECM) of the brain is essential for homeostasis and normal functions, but is rapidly remodelled during acute brain injury alongside the development of an inflammatory response driven by the cytokine interleukin (IL)-1. Whether the ECM regulates IL-1 actions in astrocytes is completely unknown. The aim of this study was to test the hypothesis that cellular attachment to the ECM is a critical mediator of IL-1beta-induced signalling pathways and development of reactive phenotype in astrocytes. Primary rat astrocytes adhered to fibronectin, laminin and fibrillin-1 in an integrin-dependent manner. Attachment to these ECM molecules significantly increased IL-1beta-induced activation of extracellular signal-regulated kinase 1/2 (ERK1/2) and inhibition of RhoA and Rho kinase (ROCK), coincident with loss of focal adhesions and cellular morphological changes. Our data demonstrate that the ECM regulates IL-1 actions in astrocytes via cross-talk mechanisms between ERK1/2 and RhoA/ROCK, which could have important implications in brain inflammatory disorders.
Figures
Similar articles
-
Activation of brain endothelial cells by interleukin-1 is regulated by the extracellular matrix after acute brain injury.Mol Cell Neurosci. 2013 Nov;57:93-103. doi: 10.1016/j.mcn.2013.10.007. Epub 2013 Oct 24. Mol Cell Neurosci. 2013. PMID: 24161715
-
Interleukin-1beta induces a reactive astroglial phenotype via deactivation of the Rho GTPase-Rock axis.J Neurosci. 2004 Mar 17;24(11):2837-45. doi: 10.1523/JNEUROSCI.4789-03.2004. J Neurosci. 2004. PMID: 15028778 Free PMC article.
-
Adhesion to fibronectin regulates interleukin-1 beta expression in microglial cells.Mol Cell Neurosci. 2009 Jun;41(2):148-55. doi: 10.1016/j.mcn.2009.02.007. Epub 2009 Feb 26. Mol Cell Neurosci. 2009. PMID: 19250967
-
The extracellular matrix protein laminin-10 promotes blood-brain barrier repair after hypoxia and inflammation in vitro.J Neuroinflammation. 2016 Feb 1;13:25. doi: 10.1186/s12974-016-0495-9. J Neuroinflammation. 2016. PMID: 26832174 Free PMC article.
-
Extracellular signal-regulated kinase promotes Rho-dependent focal adhesion formation by suppressing p190A RhoGAP.Mol Cell Biol. 2010 Jul;30(13):3233-48. doi: 10.1128/MCB.01178-09. Epub 2010 May 3. Mol Cell Biol. 2010. PMID: 20439493 Free PMC article.
Cited by
-
LPS and IL-1 differentially activate mouse and human astrocytes: role of CD14.Glia. 2014 Jun;62(6):999-1013. doi: 10.1002/glia.22657. Epub 2014 Mar 21. Glia. 2014. PMID: 24659539 Free PMC article.
-
Interleukin-1β Enhances Umbilical Cord Mesenchymal Stem Cell Adhesion Ability on Human Umbilical Vein Endothelial Cells via LFA-1/ICAM-1 Interaction.Stem Cells Int. 2019 Dec 27;2019:7267142. doi: 10.1155/2019/7267142. eCollection 2019. Stem Cells Int. 2019. PMID: 31949440 Free PMC article.
-
The Biology of Regeneration Failure and Success After Spinal Cord Injury.Physiol Rev. 2018 Apr 1;98(2):881-917. doi: 10.1152/physrev.00017.2017. Physiol Rev. 2018. PMID: 29513146 Free PMC article. Review.
-
TIMP-1 couples RhoK activation to IL-1β-induced astrocyte responses.Neurosci Lett. 2015 Nov 16;609:165-70. doi: 10.1016/j.neulet.2015.10.038. Epub 2015 Oct 17. Neurosci Lett. 2015. PMID: 26484505 Free PMC article.
-
The biology of interleukin-1: emerging concepts in the regulation of the actin cytoskeleton and cell junction dynamics.Cell Mol Life Sci. 2012 Feb;69(4):487-500. doi: 10.1007/s00018-011-0760-0. Epub 2011 Jul 9. Cell Mol Life Sci. 2012. PMID: 21744066 Free PMC article. Review.
References
-
- Bellail A.C., Hunter S.B., Brat D.J., Tan C., Van Meir E.G. Microregional extracellular matrix heterogeneity in brain modulates glioma cell invasion. Int. J. Biochem. Cell Biol. 2004;36:1046–1069. - PubMed
-
- Cain S.A., Baldock C., Gallagher J., Morgan A., Bax D.V., Weiss A.S., Shuttleworth C.A., Kielty C.M. Fibrillin-1 interactions with heparin: implications for microfibril and elastic fiber assembly. J. Biol. Chem. 2005;280:30526–30537. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
