

Mucopolysaccharidosis VI
- PMID: 20385007
- PMCID: PMC2873242
- DOI: 10.1186/1750-1172-5-5
Mucopolysaccharidosis VI
Abstract
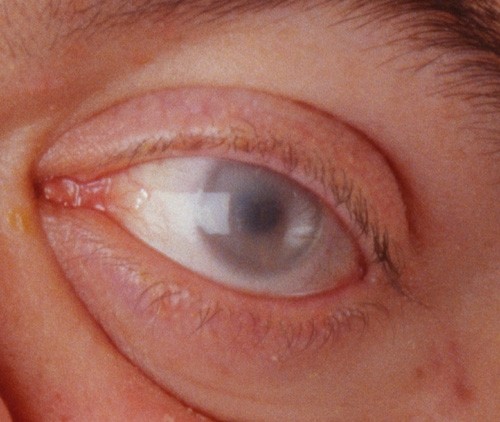
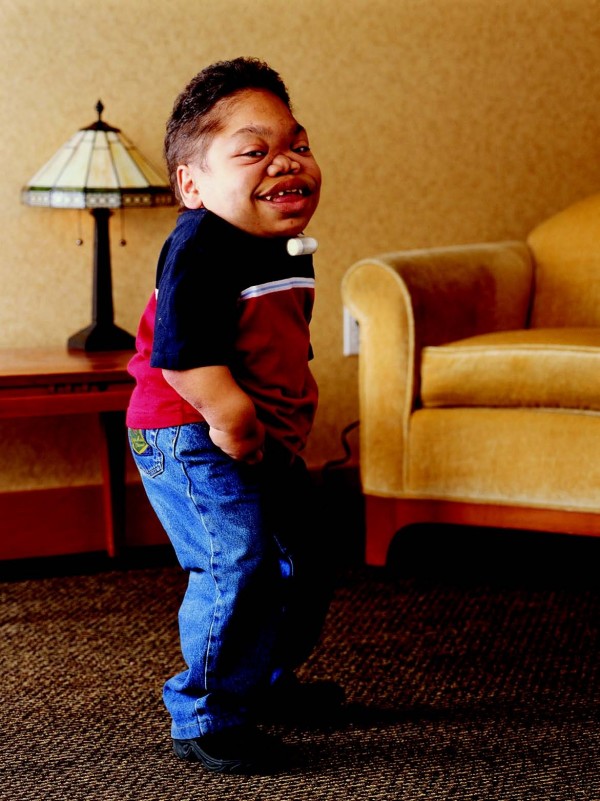
Mucopolysaccharidosis VI (MPS VI) is a lysosomal storage disease with progressive multisystem involvement, associated with a deficiency of arylsulfatase B leading to the accumulation of dermatan sulfate. Birth prevalence is between 1 in 43,261 and 1 in 1,505,160 live births. The disorder shows a wide spectrum of symptoms from slowly to rapidly progressing forms. The characteristic skeletal dysplasia includes short stature, dysostosis multiplex and degenerative joint disease. Rapidly progressing forms may have onset from birth, elevated urinary glycosaminoglycans (generally >100 microg/mg creatinine), severe dysostosis multiplex, short stature, and death before the 2nd or 3rd decades. A more slowly progressing form has been described as having later onset, mildly elevated glycosaminoglycans (generally <100 microg/mg creatinine), mild dysostosis multiplex, with death in the 4th or 5th decades. Other clinical findings may include cardiac valve disease, reduced pulmonary function, hepatosplenomegaly, sinusitis, otitis media, hearing loss, sleep apnea, corneal clouding, carpal tunnel disease, and inguinal or umbilical hernia. Although intellectual deficit is generally absent in MPS VI, central nervous system findings may include cervical cord compression caused by cervical spinal instability, meningeal thickening and/or bony stenosis, communicating hydrocephalus, optic nerve atrophy and blindness. The disorder is transmitted in an autosomal recessive manner and is caused by mutations in the ARSB gene, located in chromosome 5 (5q13-5q14). Over 130 ARSB mutations have been reported, causing absent or reduced arylsulfatase B (N-acetylgalactosamine 4-sulfatase) activity and interrupted dermatan sulfate and chondroitin sulfate degradation. Diagnosis generally requires evidence of clinical phenotype, arylsulfatase B enzyme activity <10% of the lower limit of normal in cultured fibroblasts or isolated leukocytes, and demonstration of a normal activity of a different sulfatase enzyme (to exclude multiple sulfatase deficiency). The finding of elevated urinary dermatan sulfate with the absence of heparan sulfate is supportive. In addition to multiple sulfatase deficiency, the differential diagnosis should also include other forms of MPS (MPS I, II IVA, VII), sialidosis and mucolipidosis. Before enzyme replacement therapy (ERT) with galsulfase (Naglazyme), clinical management was limited to supportive care and hematopoietic stem cell transplantation. Galsulfase is now widely available and is a specific therapy providing improved endurance with an acceptable safety profile. Prognosis is variable depending on the age of onset, rate of disease progression, age at initiation of ERT and on the quality of the medical care provided.
Figures










References
-
- Maroteaux P, Leveque B, Marie J, Lamy M. A new dysostosis with urinary elimination of chondroitin sulfate B. Presse Med. 1963;71:1849–1852. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
