

RGS4 is a negative regulator of insulin release from pancreatic beta-cells in vitro and in vivo
- PMID: 20385802
- PMCID: PMC2867926
- DOI: 10.1073/pnas.1003655107
RGS4 is a negative regulator of insulin release from pancreatic beta-cells in vitro and in vivo
Abstract
Therapeutic strategies that augment insulin release from pancreatic beta-cells are considered beneficial in the treatment of type 2 diabetes. We previously demonstrated that activation of beta-cell M(3) muscarinic receptors (M3Rs) greatly promotes glucose-stimulated insulin secretion (GSIS), suggesting that strategies aimed at enhancing signaling through beta-cell M3Rs may become therapeutically useful. M3R activation leads to the stimulation of G proteins of the G(q) family, which are under the inhibitory control of proteins known as regulators of G protein signaling (RGS proteins). At present, it remains unknown whether RGS proteins play a role in regulating insulin release. To address this issue, we initially demonstrated that MIN6 insulinoma cells express functional M3Rs and that RGS4 was by far the most abundant RGS protein expressed by these cells. Strikingly, siRNA-mediated knockdown of RGS4 expression in MIN6 cells greatly enhanced M3R-mediated augmentation of GSIS and calcium release. We obtained similar findings using pancreatic islets prepared from RGS4-deficient mice. Interestingly, RGS4 deficiency had little effect on insulin release caused by activation of other beta-cell GPCRs. Finally, treatment of mutant mice selectively lacking RGS4 in pancreatic beta-cells with a muscarinic agonist (bethanechol) led to significantly increased plasma insulin and reduced blood glucose levels, as compared to control littermates. Studies with beta-cell-specific M3R knockout mice showed that these responses were mediated by beta-cell M3Rs. These findings indicate that RGS4 is a potent negative regulator of M3R function in pancreatic beta-cells, suggesting that RGS4 may represent a potential target to promote insulin release for therapeutic purposes.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
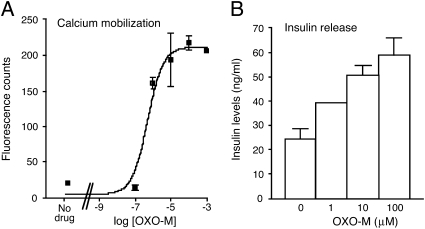
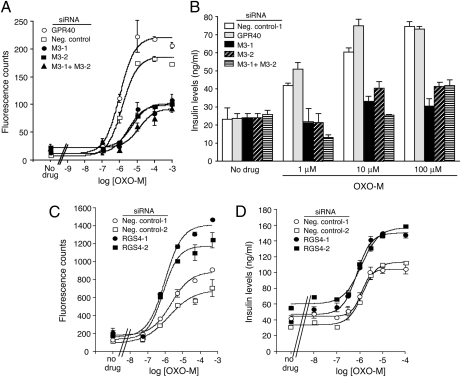

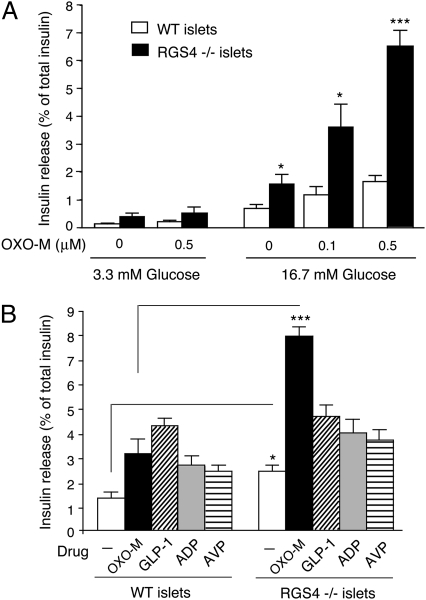
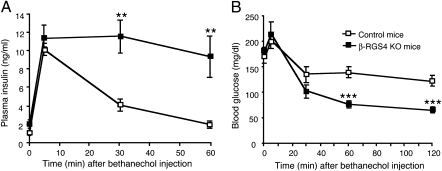
References
-
- Kahn SE. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia. 2003;46:3–19. - PubMed
-
- Moller DE. New drug targets for type 2 diabetes and the metabolic syndrome. Nature. 2001;414:821–827. - PubMed
-
- Ahrén B. Islet G protein-coupled receptors as potential targets for treatment of type 2 diabetes. Nat Rev Drug Discov. 2009;8:369–385. - PubMed
-
- Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639–650. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
