

Global analysis of gene activity during Arabidopsis seed development and identification of seed-specific transcription factors
- PMID: 20385809
- PMCID: PMC2889569
- DOI: 10.1073/pnas.1003530107
Global analysis of gene activity during Arabidopsis seed development and identification of seed-specific transcription factors
Abstract
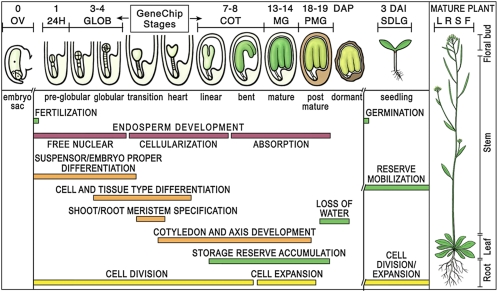
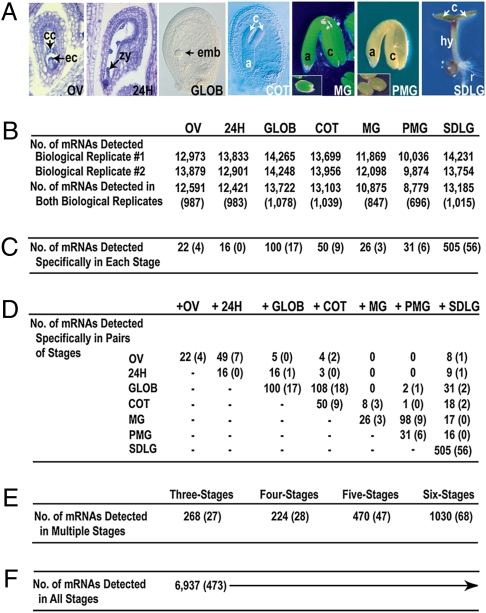
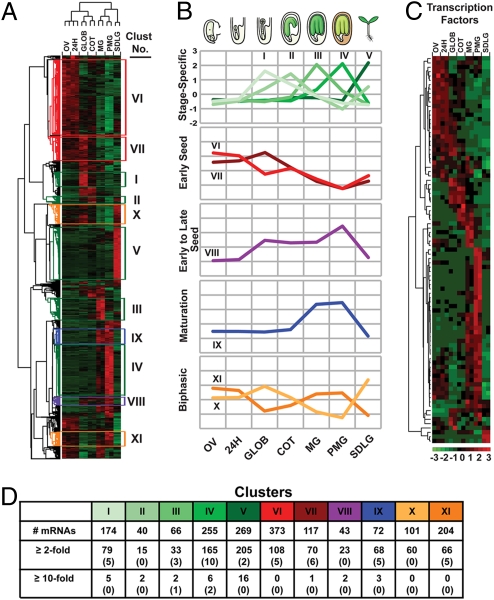
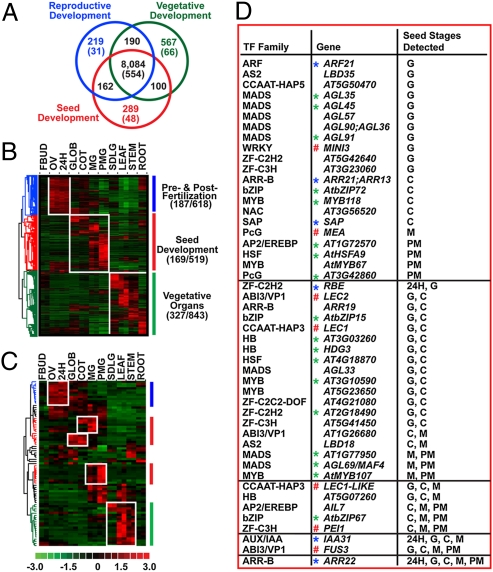
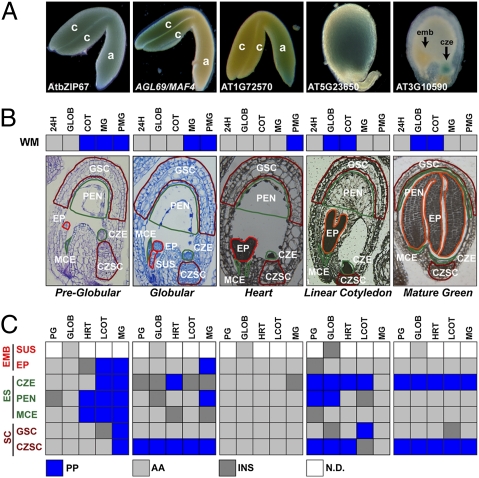
Most of the transcription factors (TFs) responsible for controlling seed development are not yet known. To identify TF genes expressed at specific stages of seed development, including those unique to seeds, we used Affymetrix GeneChips to profile Arabidopsis genes active in seeds from fertilization through maturation and at other times of the plant life cycle. Seed gene sets were compared with those expressed in prefertilization ovules, germinating seedlings, and leaves, roots, stems, and floral buds of the mature plant. Most genes active in seeds are shared by all stages of seed development, although significant quantitative changes in gene activity occur. Each stage of seed development has a small gene set that is either specific at the level of the GeneChip or up-regulated with respect to genes active at other stages, including those that encode TFs. We identified 289 seed-specific genes, including 48 that encode TFs. Seven of the seed-specific TF genes are known regulators of seed development and include the LEAFY COTYLEDON (LEC) genes LEC1, LEC1-LIKE, LEC2, and FUS3. The rest represent different classes of TFs with unknown roles in seed development. Promoter-beta-glucuronidase (GUS) fusion experiments and seed mRNA localization GeneChip datasets showed that the seed-specific TF genes are active in different compartments and tissues of the seed at unique times of development. Collectively, these seed-specific TF genes should facilitate the identification of regulatory networks that are important for programming seed development.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Goldberg RB, de Paiva G, Yadegari R. Plant embryogenesis: Zygote to seed. Science. 1994;266:605–614. - PubMed
-
- Raghavan V. Double Fertilization: Embryo and Endosperm Development in Flowering Plants. Berlin: Springer; 2006.
-
- Goldberg RB, Barker SJ, Perez-Grau L. Regulation of gene expression during plant embryogenesis. Cell. 1989;56:149–160. - PubMed
-
- Harada JJ. In: Seed maturation and the control of germination. Cellular and Molecular Biology of Seed Development, Advances in Cellular and Molecular Biology of Plants. Larkins BA, Vasil IK, editors. Vol. 4. Dordrecht, the Netherlands: Kluwer Academic Publishers; 1997. pp. 545–592.
-
- Baud S, Boutin J-P, Miquel M, Lepiniec L, Rochat C. Anintegrated overview of seed development in Arabidopsis thaliana ecotype WS. Plant Physiol Biochem. 2002;40:151–160.
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
