

On the conservation of the slow conformational dynamics within the amino acid kinase family: NAGK the paradigm
- PMID: 20386738
- PMCID: PMC2851564
- DOI: 10.1371/journal.pcbi.1000738
On the conservation of the slow conformational dynamics within the amino acid kinase family: NAGK the paradigm
Abstract
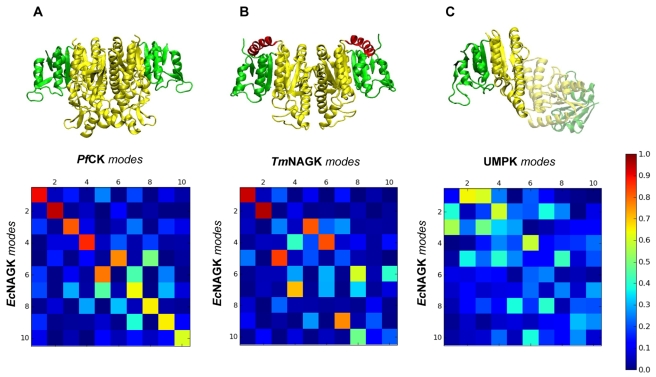
N-acetyl-L-glutamate kinase (NAGK) is the structural paradigm for examining the catalytic mechanisms and dynamics of amino acid kinase family members. Given that the slow conformational dynamics of the NAGK (at the microseconds time scale or slower) may be rate-limiting, it is of importance to assess the mechanisms of the most cooperative modes of motion intrinsically accessible to this enzyme. Here, we present the results from normal mode analysis using an elastic network model representation, which shows that the conformational mechanisms for substrate binding by NAGK strongly correlate with the intrinsic dynamics of the enzyme in the unbound form. We further analyzed the potential mechanisms of allosteric signalling within NAGK using a Markov model for network communication. Comparative analysis of the dynamics of family members strongly suggests that the low-frequency modes of motion and the associated intramolecular couplings that establish signal transduction are highly conserved among family members, in support of the paradigm sequence-->structure-->dynamics-->function.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
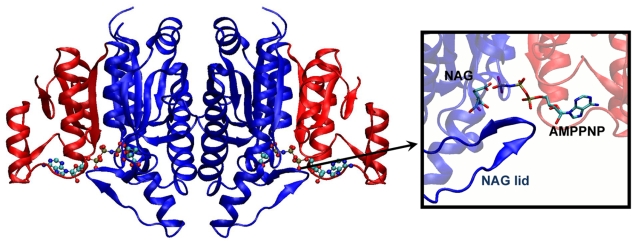
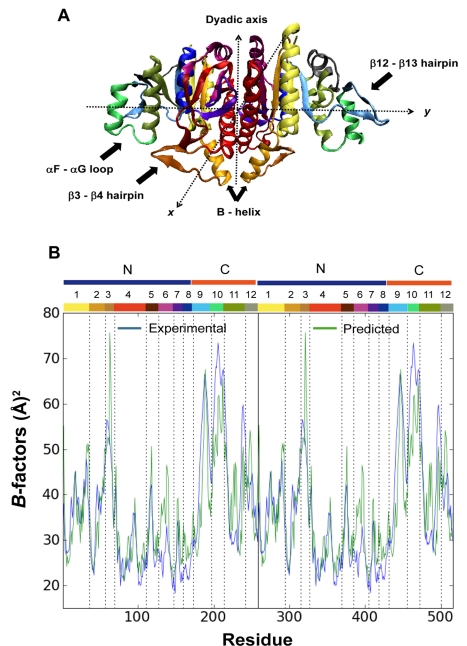
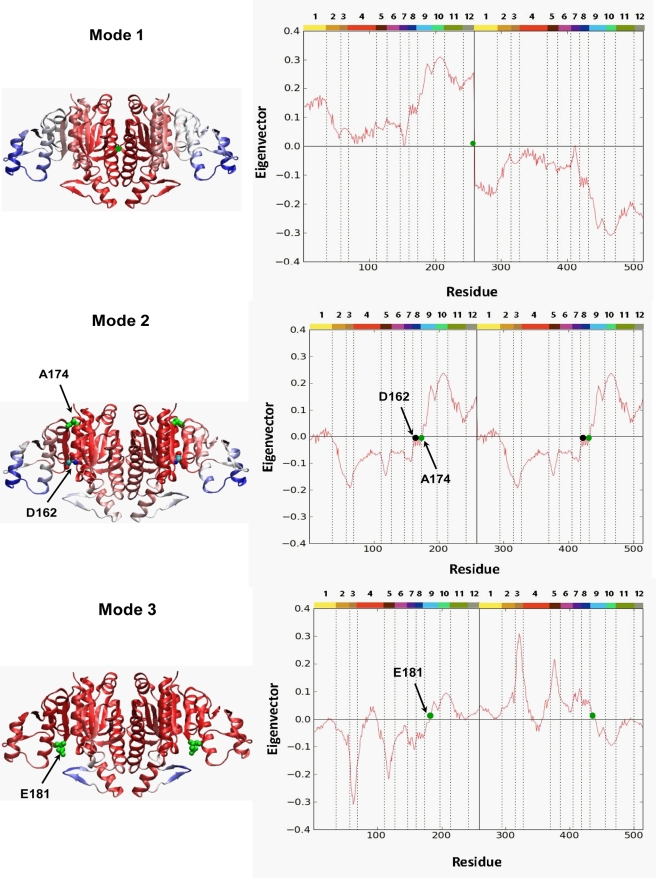
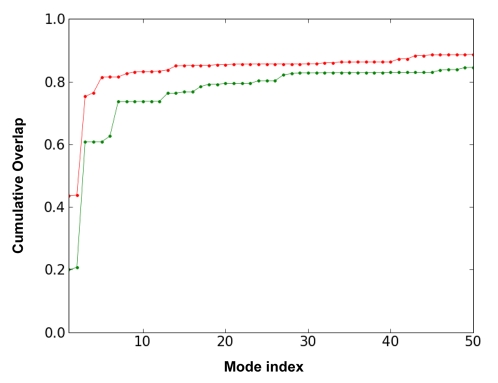
 ) (equation (8) in
Methods
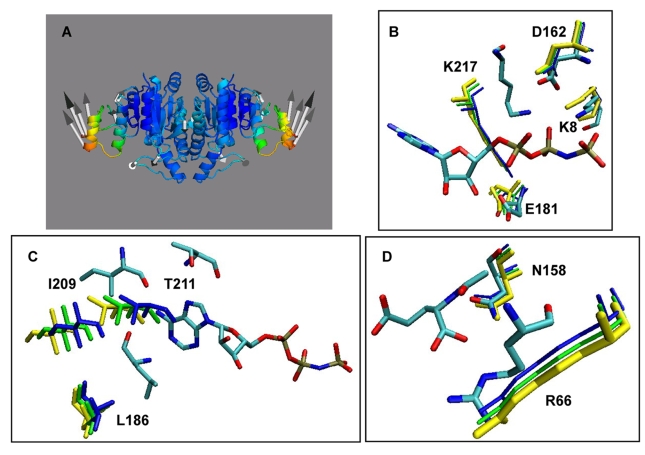
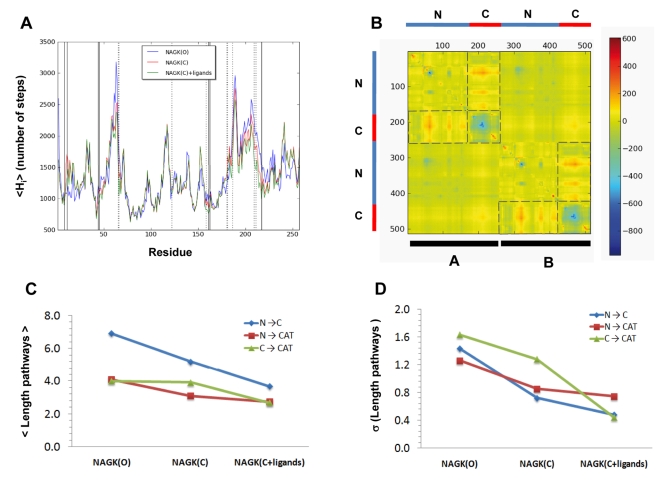
) of the open and ligand-bound closed forms. Dashed lines set the boundaries of N- and C- domains and also enclose those pairs of domains that undergo the largest changes in the contribution from cross-correlations upon ligand binding. (C) Mean path lengths for linking different parts of the protein: N- and C-domains (blue), N-domain and catalytic site (red), and C-domain and catalytic site (green). (D) Standard deviation in the mean paths displayed in panel (C).
) (equation (8) in
Methods
) of the open and ligand-bound closed forms. Dashed lines set the boundaries of N- and C- domains and also enclose those pairs of domains that undergo the largest changes in the contribution from cross-correlations upon ligand binding. (C) Mean path lengths for linking different parts of the protein: N- and C-domains (blue), N-domain and catalytic site (red), and C-domain and catalytic site (green). (D) Standard deviation in the mean paths displayed in panel (C).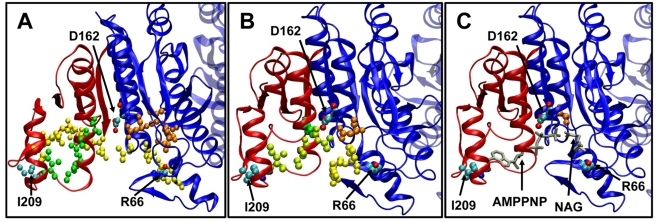
Similar articles
-
Changes in dynamics upon oligomerization regulate substrate binding and allostery in amino acid kinase family members.PLoS Comput Biol. 2011 Sep;7(9):e1002201. doi: 10.1371/journal.pcbi.1002201. Epub 2011 Sep 29. PLoS Comput Biol. 2011. PMID: 21980279 Free PMC article.
-
Conformational dynamics play important roles upon the function of N-acetylglutamate kinase.Appl Microbiol Biotechnol. 2017 May;101(9):3485-3492. doi: 10.1007/s00253-017-8237-1. Epub 2017 Mar 24. Appl Microbiol Biotechnol. 2017. PMID: 28341883 Review.
-
Site-directed mutagenesis of Escherichia coli acetylglutamate kinase and aspartokinase III probes the catalytic and substrate-binding mechanisms of these amino acid kinase family enzymes and allows three-dimensional modelling of aspartokinase.J Mol Biol. 2003 Nov 28;334(3):459-76. doi: 10.1016/j.jmb.2003.09.038. J Mol Biol. 2003. PMID: 14623187
-
Phosphorylation Mechanism of N-Acetyl-l-glutamate Kinase, a QM/MM Study.J Phys Chem B. 2019 Apr 4;123(13):2844-2852. doi: 10.1021/acs.jpcb.9b00547. Epub 2019 Mar 21. J Phys Chem B. 2019. PMID: 30848915
-
Arginine and nitrogen storage.Curr Opin Struct Biol. 2008 Dec;18(6):673-81. doi: 10.1016/j.sbi.2008.11.002. Epub 2008 Nov 27. Curr Opin Struct Biol. 2008. PMID: 19013524 Review.
Cited by
-
A phylogenetic analysis of normal modes evolution in enzymes and its relationship to enzyme function.J Mol Biol. 2012 Sep 21;422(3):442-59. doi: 10.1016/j.jmb.2012.05.028. Epub 2012 May 28. J Mol Biol. 2012. PMID: 22651983 Free PMC article.
-
Comparison of the Internal Dynamics of Metalloproteases Provides New Insights on Their Function and Evolution.PLoS One. 2015 Sep 23;10(9):e0138118. doi: 10.1371/journal.pone.0138118. eCollection 2015. PLoS One. 2015. PMID: 26397984 Free PMC article.
-
The underappreciated role of allostery in the cellular network.Annu Rev Biophys. 2013;42:169-89. doi: 10.1146/annurev-biophys-083012-130257. Epub 2013 Feb 28. Annu Rev Biophys. 2013. PMID: 23451894 Free PMC article. Review.
-
The crystal structure of Thermus thermophilus UMP kinase complexed with a phosphoryl group acceptor and donor.PLoS One. 2025 Sep 2;20(9):e0330398. doi: 10.1371/journal.pone.0330398. eCollection 2025. PLoS One. 2025. PMID: 40892736 Free PMC article.
-
FindTargetsWEB: A User-Friendly Tool for Identification of Potential Therapeutic Targets in Metabolic Networks of Bacteria.Front Genet. 2019 Jul 4;10:633. doi: 10.3389/fgene.2019.00633. eCollection 2019. Front Genet. 2019. PMID: 31333719 Free PMC article.
References
-
- Changeux JP, Edelstein SJ. Allosteric mechanisms of signal transduction. Science. 2005;308:1424–1428. - PubMed
-
- Henzler-Wildman K, Kern D. Dynamic personalities of proteins. Nature. 2007;450:964–972. - PubMed
-
- Lange OF, Lakomek NA, Fares C, Schroder GF, Walter KFA, et al. Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science. 2008;320:1471–1475. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
