

Identification and characterization of eight novel SMPD1 mutations causing types A and B Niemann-Pick disease
- PMID: 20386867
- PMCID: PMC2896470
- DOI: 10.2119/molmed.2010.00017
Identification and characterization of eight novel SMPD1 mutations causing types A and B Niemann-Pick disease
Abstract
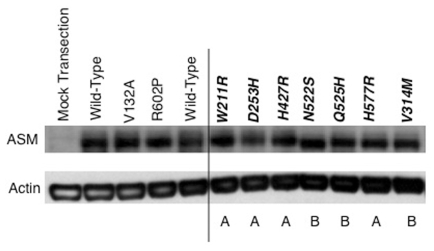
Types A and B Niemann-Pick disease (NPD) result from the deficient activity of acid sphingomyelinase (ASM), due to mutations in the sphingomyelin phosphodiesterase 1 (SMPD1) gene. Here we report the identification, characterization and genotype/phenotype correlations of eight novel mutations in six unrelated NPD patients. These mutations included seven missense mutations: c.631T>C (p.W211R), c.757G>C (p.D253H), c.940G>A (p.V314M), c.1280A>G (p.H427R), c.1564A>G (p.N522S), c.1575G>C (p.Q525H) and c.1729A>G (p.H577R), and a novel frameshift mutation, c.1657delACCGCCT (fsT553). Each missense mutation was expressed in 293T or COS-7 cells; mutant enzymes p.W211R, p.D253H, p.H427R and p.H577R had <1% of expressed wild-type activity, whereas p.V314M, p.N522S and p.Q525H had 21.7%, 10.1% and 64% of expressed wild-type activity, respectively. The c.1564A>G mutation obliterated a known N-glycosylation site and its p.N522S mutant enzyme had ~10% of expressed wild-type activity. Western blot analysis revealed that each mutant protein was expressed at near wild-type amounts, despite their differences in residual activity. The novel seven-base deletion occurred at codon 553, leading to a premature truncation after residue 609. The expression studies predicted the clinical phenotypes of the six patients: two type A patients had genotypes with only type A alleles [c.631T>C (p.W211R), c.757G>C (p.D253H) and c.1729A>G (p.H577R)], and the other four type B disease patients had at least one neuroprotective mutant type B allele [c.940G>A (p.V314M), c.1280A>G (p.H427R), c.1564A>G (p.N522S) and c.1575G>C (p.Q525H)] that expressed >5% residual ASM activity. Thus, these new mutations provide novel genotype/phenotype correlations and further document the genetic heterogeneity in types A and B NPD.
Figures
References
-
- Schuchman EH. The pathogenesis and treatment of acid sphingomyelinase-deficient Niemann-Pick disease. J. Inherit. Metab. Dis. 2007;30:654–63. - PubMed
-
- Patterson MC, et al. Niemann-Pick disease type C: a lipid trafficking disorder. In: Scriver CR, et al., editors. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. McGraw-Hill; New York: 2001. pp. 3611–34.
-
- Ferlinz K, et al. Functional characterization of the N-glycosylation sites of human acid sphingomyelinase by site-directed mutagenesis. Eur. J. Biochem. 1997;243:511–7. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
