

Review
doi: 10.1093/hmg/ddq142.
Epub 2010 Apr 13.
Current status on Alzheimer disease molecular genetics: from past, to present, to future
Affiliations
- PMID: 20388643
- PMCID: PMC2875058
- DOI: 10.1093/hmg/ddq142
Item in Clipboard
Review
Current status on Alzheimer disease molecular genetics: from past, to present, to future
Hum Mol Genet.
.
Abstract
Linkage studies, candidate gene and whole-genome association studies have resulted in a tremendous amount of putative risk genes for Alzheimer's disease (AD). Yet, besides the three causal genes-amyloid precursor protein and presenilin 1 and 2 genes-and one risk gene apolipoprotein E (APOE), no single functional risk variant was identified. Discussing the possible involvement of rare alleles and other types of genetic variants, this review summarizes the current knowledge on the genetic spectrum of AD and integrates different approaches and recent discoveries by genome-wide association studies.
Figures
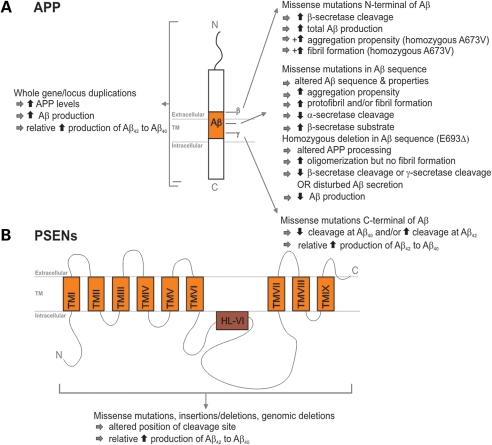
Effect of different causal APP and PSEN1 mutations on APP processing and Aβ generation. (A) Schematic presentation of the APP protein structure. Right, the effect of APP mutations on APP processing is given according to their location relative to the Aβ peptide. The additional effects of the N-terminal recessive mutations A673V and E693Δ are indicated by the ‘+’ symbol. Left, the effect of whole APP gene or locus duplications is depicted. (B) Schematic presentation of the PSEN protein structure. Boxes represent the transmembrane regions that are separated by hydrophilic loops. The effect of different mutations scattered throughout the protein is summarized.
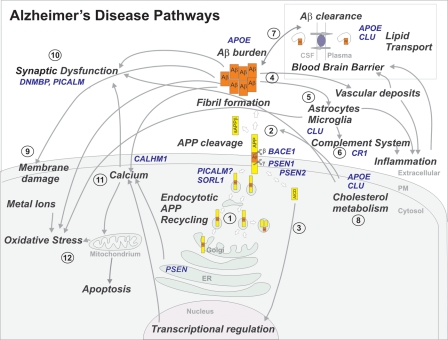
Overview of several disease pathways involved in AD pathogenesis. Causal AD genes and AD risk factors are marked in blue. APP is synthesized by the endoplasmatic reticulum (ER) and the Golgi apparatus (1). Following the amyloidogenic pathway in neurons, APP is cleaved by β-secretase (BACE1) and γ-secretase (PSEN) to generate Aβ peptides and the amyloid intracellular domain [AICD] (2), which influences the transcription of several genes (3). In the APP retromer recycling pathway (1), APP is redirected to endosomes by SORL1. PICALM has a presumed role in APP endocytotic recycling (1). Aβ monomers aggregate into Aβ fibrils, causing amyloid plaques in brain parenchyma and vasculature (4). Aβ activates microglia and astrocytes, inducing the complement system, local inflammatory responses and oxidative stress (5). CR1 is the receptor of the complement C3b protein and participates in the clearance of Aβ from circulation (6). Besides causing increased Aβ endocytosis into glial cells, CLU is involved in Aβ clearance at the blood–brain barrier (7). APOE enhances amyloid plaque formation by conformational changes of Aβ. Clusterin (APOJ) and APOE are the main escorting proteins of Aβ in brain (7). Both are also important in cholesterol metabolism at the neuronal membrane (8) and high intracellular cholesterol may enhance APP amyloidogenic processing (2), which in turn can lead to membrane damage (9). Moreover, impaired cholesterol metabolism may influence synaptic dysfunction (10). Both PICALM and DNMBP are related at the synapse (10). Interaction of Aβ oligomers at the membrane is further connected to the calcium hypothesis in AD (11). Polymorphisms in the Ca2+ channel CALHM1 impair Ca2+ permeability at the plasma membrane (11). In addition, PSENs function as ER Ca2+-leak channels and several early-onset mutations impair Ca2+-leak-channel function, resulting in an excessive Ca2+ accumulation in the cytosol. An excessive Ca2+ is taken up by mitochondria, further leading to oxidative stress and apoptosis (12).
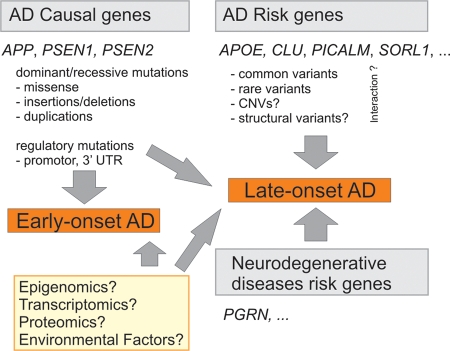
Schematic summary of the current knowledge of AD molecular genetics. Mutations in causal AD genes APP, PSEN1 and PSEN2 are associated with early-onset AD. Other mutations such as regulatory variants in promoter regions or 3′ untranslated regions (3′-UTR) of causal AD genes may confer susceptibility to late-onset AD. Multiple common and/or rare variants in AD susceptibility genes confer risk to late-onset AD. The top four risk genes (Alzforum, status on 2 April 2010) are listed. The role of CNVs and other structural variants in late-onset AD remains to be clarified, likewise for the interaction between the different risk factors (indicated by question marks). Genes associated with other neurodegenerative dementias like PGRN are involved in AD genetic etiology as well. Several different approaches will likely reveal new genes involved in early and late-onset AD (yellow box).
References
-
- Alzheimer's Disease International Consortium. AD International, World Alzheimer Report. 2009. Alzheimer's disease International, London. Available at http://www.alz.co.uk/
-
- Lobo A., Launer L.J., Fratiglioni L., Andersen K., Di Carlo A., Breteler M.M., Copeland J.R., Dartigues J.F., Jagger C., Martinez-Lage J., et al. Prevalence of dementia and major subtypes in Europe: a collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology. 2000;54:S4–S9. - PubMed
-
- McKhann G., Drachman D., Folstein M., Katzman R., Price D., Stadlan E.M. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–944. - PubMed
-
- van der Zee J., Sleegers K., Van Broeckhoven C. Invited article: the AD-frontotemporal lobar degeneration spectrum. Neurology. 2008;71:1191–1197. doi:10.1212/01.wnl.0000327523.52537.86. - DOI - PubMed
-
- Braak H., Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi:10.1007/BF00308809. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
