

Genetic associations in type I interferon related pathways with autoimmunity
- PMID: 20392289
- PMCID: PMC2991775
- DOI: 10.1186/ar2883
Genetic associations in type I interferon related pathways with autoimmunity
Abstract
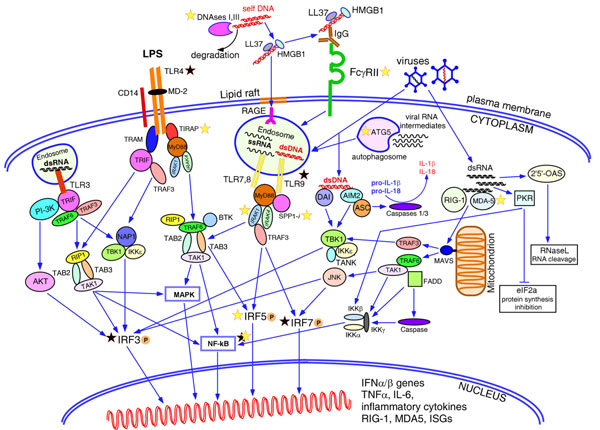
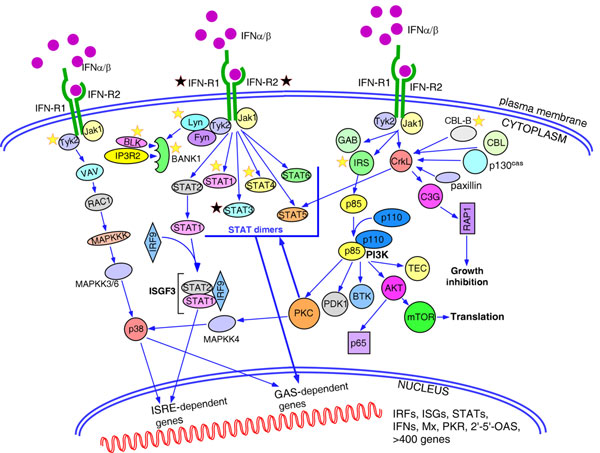
Type I interferons play an outstanding role in innate and adaptive immunity by enhancing functions of dendritic cells, inducing differentiation of monocytes, promoting immunoglobulin class switching in B cells and stimulating effector functions of T cells. The increased production of IFNα/β by plasmacytoid dendritic cells could be responsible for not only efficient antiviral defence, but it also may be a pathological factor in the development of various autoimmune disorders. The first evidence of a genetic link between type I interferons and autoimmune diseases was the observation that elevated IFNα activity is frequently detected in the sera of patients with systemic lupus erythematosus, and that this trait shows high heritability and familial aggregation in their first-degree healthy relatives. To date, a number of genes involved in interferon signalling have been associated with various autoimmune diseases. Patients with systemic lupus erythematosus, Sjögren's syndrome, dermatomyositis, psoriasis, and a fraction of patients with rheumatoid arthritis display a specific expression pattern of interferon-dependent genes in their leukocytes, termed the interferon signature. Here, in an attempt to understand the role of type I interferons in the pathogenesis of autoimmunity, we review the recent advances in the genetics of autoimmune diseases focusing on the association of genes involved in type I interferon pathways.
Figures
Similar articles
-
Expression of Long Interspersed Nuclear Element 1 Retroelements and Induction of Type I Interferon in Patients With Systemic Autoimmune Disease.Arthritis Rheumatol. 2016 Nov;68(11):2686-2696. doi: 10.1002/art.39795. Arthritis Rheumatol. 2016. PMID: 27338297 Free PMC article.
-
Interferon regulatory factors: beyond the antiviral response and their link to the development of autoimmune pathology.Autoimmun Rev. 2011 Dec;11(2):98-103. doi: 10.1016/j.autrev.2011.08.006. Epub 2011 Aug 18. Autoimmun Rev. 2011. PMID: 21872684 Review.
-
Functionally impaired plasmacytoid dendritic cells and non-haematopoietic sources of type I interferon characterize human autoimmunity.Nat Commun. 2020 Dec 1;11(1):6149. doi: 10.1038/s41467-020-19918-z. Nat Commun. 2020. PMID: 33262343 Free PMC article.
-
Type I Interferon and the Spectrum of Susceptibility to Viral Infection and Autoimmune Disease: A Shared Genomic Signature.Front Immunol. 2021 Nov 30;12:757249. doi: 10.3389/fimmu.2021.757249. eCollection 2021. Front Immunol. 2021. PMID: 34917078 Free PMC article. Review.
-
Fueling autoimmunity: type I interferon in autoimmune diseases.Expert Rev Clin Immunol. 2013 Mar;9(3):201-10. doi: 10.1586/eci.12.106. Expert Rev Clin Immunol. 2013. PMID: 23445195 Free PMC article. Review.
Cited by
-
Twins discordant for myositis and systemic lupus erythematosus show markedly enriched autoantibodies in the affected twin supporting environmental influences in pathogenesis.BMC Musculoskelet Disord. 2014 Mar 6;15:67. doi: 10.1186/1471-2474-15-67. BMC Musculoskelet Disord. 2014. PMID: 24602337 Free PMC article.
-
Baricitinib therapy response in rheumatoid arthritis patients associates to STAT1 phosphorylation in monocytes.Front Immunol. 2022 Jul 25;13:932240. doi: 10.3389/fimmu.2022.932240. eCollection 2022. Front Immunol. 2022. PMID: 35958600 Free PMC article.
-
Immune response biomarkers in human and veterinary research.Comp Immunol Microbiol Infect Dis. 2018 Aug;59:57-62. doi: 10.1016/j.cimid.2018.09.008. Epub 2018 Sep 24. Comp Immunol Microbiol Infect Dis. 2018. PMID: 30290889 Free PMC article. Review.
-
Pathogen recognition receptor signaling accelerates phosphorylation-dependent degradation of IFNAR1.PLoS Pathog. 2011 Jun;7(6):e1002065. doi: 10.1371/journal.ppat.1002065. Epub 2011 Jun 9. PLoS Pathog. 2011. PMID: 21695243 Free PMC article.
-
C1q-mediated repression of human monocytes is regulated by leukocyte-associated Ig-like receptor 1 (LAIR-1).Mol Med. 2015 Feb 5;20(1):559-68. doi: 10.2119/molmed.2014.00185. Mol Med. 2015. PMID: 25247291 Free PMC article.
References
-
- Luft T, Pang KC, Thomas E, Hertzog P, Hart DN, Trapani J, Cebon J. Type I IFNs enhance the terminal differentiation of dendritic cells. J Immunol. 1998;161:1947–1953. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
