

Spinal muscular atrophy: mechanisms and therapeutic strategies
- PMID: 20392710
- PMCID: PMC2875050
- DOI: 10.1093/hmg/ddq147
Spinal muscular atrophy: mechanisms and therapeutic strategies
Abstract
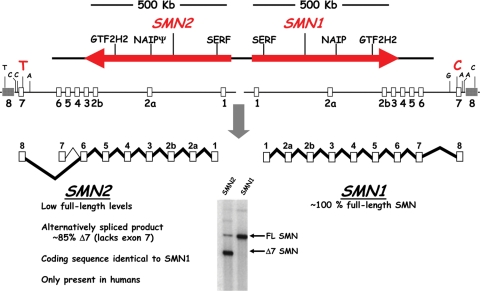
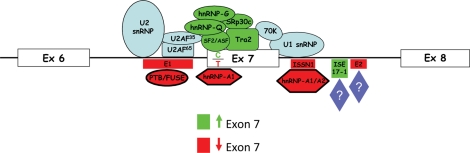
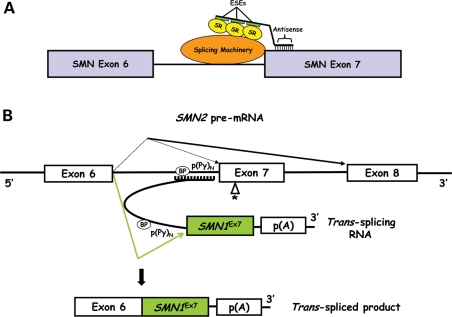
Spinal muscular atrophy (SMA) is an autosomal recessive neurodegenerative disorder and a leading genetic cause of infantile mortality. SMA is caused by mutation or deletion of Survival Motor Neuron-1 (SMN1). The clinical features of the disease are caused by specific degeneration of alpha-motor neurons in the spinal cord, leading to muscle weakness, atrophy and, in the majority of cases, premature death. A highly homologous copy gene (SMN2) is retained in almost all SMA patients but fails to generate adequate levels of SMN protein due to its defective splicing pattern. The severity of the SMA phenotype is inversely correlated with SMN2 copy number and the level of full-length SMN protein produced by SMN2 ( approximately 10-15% compared with SMN1). The natural history of SMA has been altered over the past several decades, primarily through supportive care measures, but an effective treatment does not presently exist. However, the common genetic etiology and recent progress in pre-clinical models suggest that SMA is well-suited for the development of therapeutic regimens. We summarize recent advances in translational research that hold promise for the progression towards clinical trials.
Figures
References
-
- Lefebvre S., Burglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M., et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi:10.1016/0092-8674(95)90460-3. - DOI - PubMed
-
- Feldkotter M., Schwarzer V., Wirth R., Wienker T.F., Wirth B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet. 2002;70:358–368. doi:10.1086/338627. - DOI - PMC - PubMed
-
- Rochette C.F., Gilbert N., Simard L.R. SMN gene duplication and the emergence of the SMN2 gene occurred in distinct hominids: SMN2 is unique to Homo sapiens. Hum. Genet. 2001;108:255–266. doi:10.1007/s004390100473. - DOI - PubMed
-
- Heier C.R., DiDonato C.J. Translational readthrough by the aminoglycoside geneticin (G418) modulates SMN stability in vitro and improves motor function in SMA mice in vivo. Hum. Mol. Genet. 2009;18:1310–1322. doi:10.1093/hmg/ddp030. - DOI - PMC - PubMed
-
- Lorson C.L., Androphy E.J. An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Hum. Mol. Genet. 2000;9:259–265. doi:10.1093/hmg/9.2.259. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
