

Protein kinase A phosphorylation activates Vpr-induced cell cycle arrest during human immunodeficiency virus type 1 infection
- PMID: 20392842
- PMCID: PMC2903295
- DOI: 10.1128/JVI.02273-09
Protein kinase A phosphorylation activates Vpr-induced cell cycle arrest during human immunodeficiency virus type 1 infection
Abstract
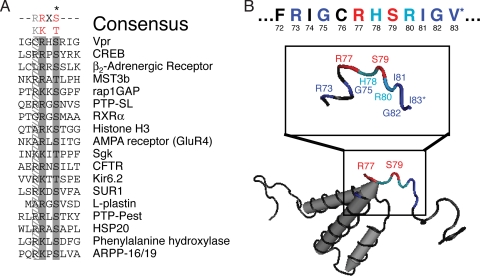
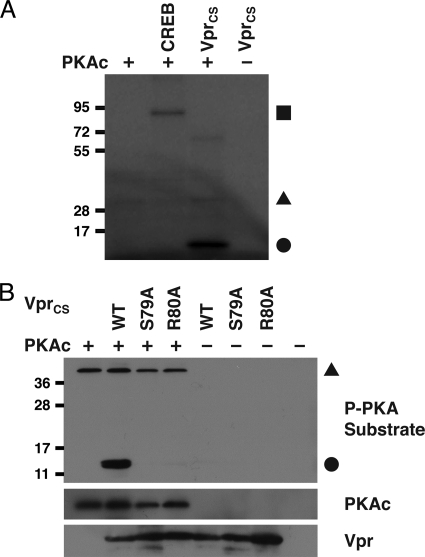
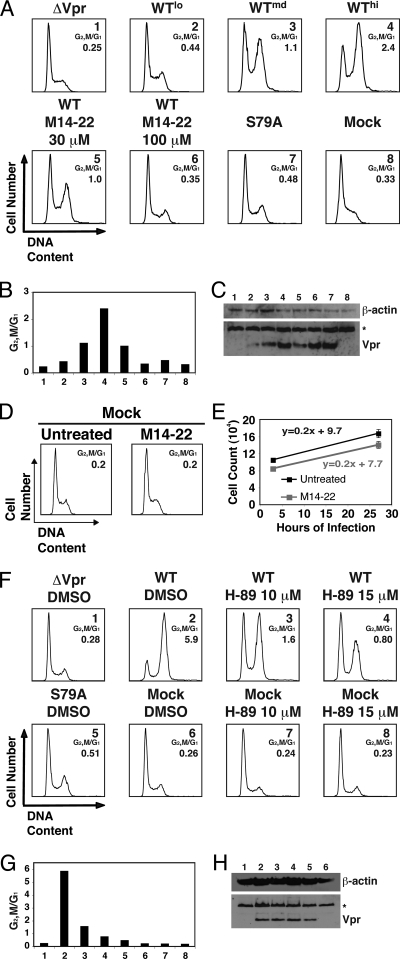
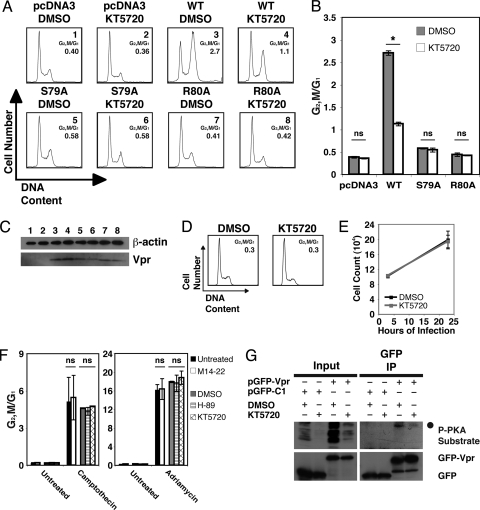
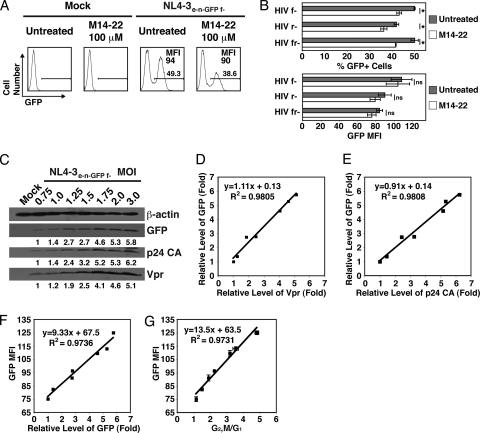
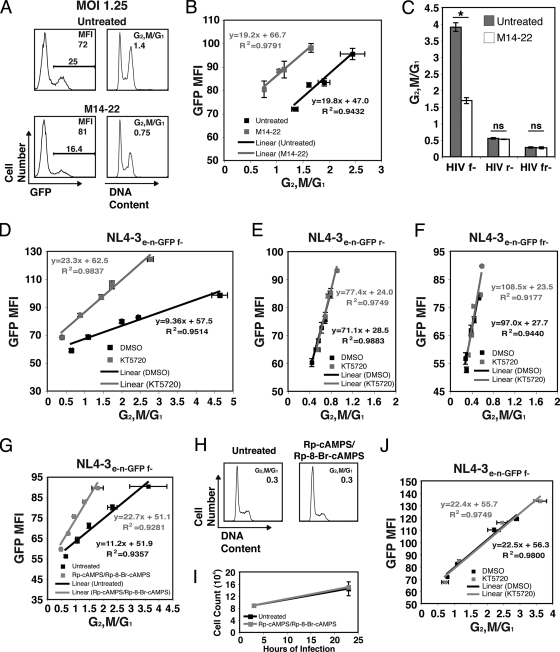
Infection with human immunodeficiency virus type 1 (HIV-1) causes an inexorable depletion of CD4(+) T cells. The loss of these cells is particularly pronounced in the mucosal immune system during acute infection, and the data suggest that direct viral cytopathicity is a major factor. Cell cycle arrest caused by the HIV-1 accessory protein Vpr is strongly correlated with virus-induced cell death, and phosphorylation of Vpr serine 79 (S79) is required to activate G(2)/M cell cycle blockade. However, the kinase responsible for phosphorylating Vpr remains unknown. Our bioinformatic analyses revealed that S79 is part of a putative phosphorylation site recognized by protein kinase A (PKA). We show here that PKA interacts with Vpr and directly phosphorylates S79. Inhibition of PKA activity during HIV-1 infection abrogates Vpr cell cycle arrest. These findings provide new insight into the signaling event that activates Vpr cell cycle arrest, ultimately leading to the death of infected T cells.
Figures



Similar articles
-
Vpr cytopathicity independent of G2/M cell cycle arrest in human immunodeficiency virus type 1-infected CD4+ T cells.J Virol. 2007 Sep;81(17):8878-90. doi: 10.1128/JVI.00122-07. Epub 2007 Jun 6. J Virol. 2007. PMID: 17553871 Free PMC article.
-
HIV-1 Vpr induces cell cycle arrest and enhances viral gene expression by depleting CCDC137.Elife. 2020 Jun 15;9:e55806. doi: 10.7554/eLife.55806. Elife. 2020. PMID: 32538781 Free PMC article.
-
The Vif and Vpr accessory proteins independently cause HIV-1-induced T cell cytopathicity and cell cycle arrest.Proc Natl Acad Sci U S A. 2006 Feb 28;103(9):3369-74. doi: 10.1073/pnas.0509417103. Epub 2006 Feb 21. Proc Natl Acad Sci U S A. 2006. PMID: 16492778 Free PMC article.
-
Human immunodeficiency virus type 1 Vpr: functions and molecular interactions.J Gen Virol. 2009 Aug;90(Pt 8):1795-1805. doi: 10.1099/vir.0.011726-0. Epub 2009 May 20. J Gen Virol. 2009. PMID: 19458171 Review.
-
Could Nef and Vpr proteins contribute to disease progression by promoting depletion of bystander cells and prolonged survival of HIV-infected cells?Biochem Biophys Res Commun. 2000 Jan 27;267(3):677-85. doi: 10.1006/bbrc.1999.1708. Biochem Biophys Res Commun. 2000. PMID: 10673351 Review.
Cited by
-
Genome-Wide Characterization of Host Transcriptional and Epigenetic Alterations During HIV Infection of T Lymphocytes.Front Immunol. 2020 Sep 10;11:2131. doi: 10.3389/fimmu.2020.02131. eCollection 2020. Front Immunol. 2020. PMID: 33013899 Free PMC article.
-
TGEV nucleocapsid protein induces cell cycle arrest and apoptosis through activation of p53 signaling.Biochem Biophys Res Commun. 2014 Mar 7;445(2):497-503. doi: 10.1016/j.bbrc.2014.02.039. Epub 2014 Feb 15. Biochem Biophys Res Commun. 2014. PMID: 24548406 Free PMC article.
-
Functional and Structural Mimicry of Cellular Protein Kinase A Anchoring Proteins by a Viral Oncoprotein.PLoS Pathog. 2016 May 3;12(5):e1005621. doi: 10.1371/journal.ppat.1005621. eCollection 2016 May. PLoS Pathog. 2016. PMID: 27137912 Free PMC article.
-
Phosphoproteomics to Characterize Host Response During Influenza A Virus Infection of Human Macrophages.Mol Cell Proteomics. 2016 Oct;15(10):3203-3219. doi: 10.1074/mcp.M116.057984. Epub 2016 Aug 2. Mol Cell Proteomics. 2016. PMID: 27486199 Free PMC article.
-
HIV-1-associated PKA acts as a cofactor for genome reverse transcription.Retrovirology. 2013 Dec 17;10:157. doi: 10.1186/1742-4690-10-157. Retrovirology. 2013. PMID: 24344931 Free PMC article.
References
-
- Adams, S. R., A. T. Harootunian, Y. J. Buechler, S. S. Taylor, and R. Y. Tsien. 1991. Fluorescence ratio imaging of cyclic AMP in single cells. Nature 349:694-697. - PubMed
-
- Agostini, I., S. Popov, T. Hao, J. H. Li, L. Dubrovsky, O. Chaika, N. Chaika, R. Lewis, and M. Bukrinsky. 2002. Phosphorylation of Vpr regulates HIV type 1 nuclear import and macrophage infection. AIDS Res. Hum. Retrovir. 18:283-288. - PubMed
-
- Altfeld, M., M. M. Addo, R. L. Eldridge, X. G. Yu, S. Thomas, A. Khatri, D. Strick, M. N. Phillips, G. B. Cohen, S. A. Islam, S. A. Kalams, C. Brander, P. J. Goulder, E. S. Rosenberg, and B. D. Walker. 2001. Vpr is preferentially targeted by CTL during HIV-1 infection. J. Immunol. 167:2743-2752. - PubMed
-
- Bachand, F., X. J. Yao, M. Hrimech, N. Rougeau, and E. A. Cohen. 1999. Incorporation of Vpr into human immunodeficiency virus type 1 requires a direct interaction with the p6 domain of the p55 gag precursor. J. Biol. Chem. 274:9083-9091. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
