

Focal cortical infarcts alter intrinsic excitability and synaptic excitation in the reticular thalamic nucleus
- PMID: 20392967
- PMCID: PMC2861582
- DOI: 10.1523/JNEUROSCI.5083-09.2010
Focal cortical infarcts alter intrinsic excitability and synaptic excitation in the reticular thalamic nucleus
Abstract
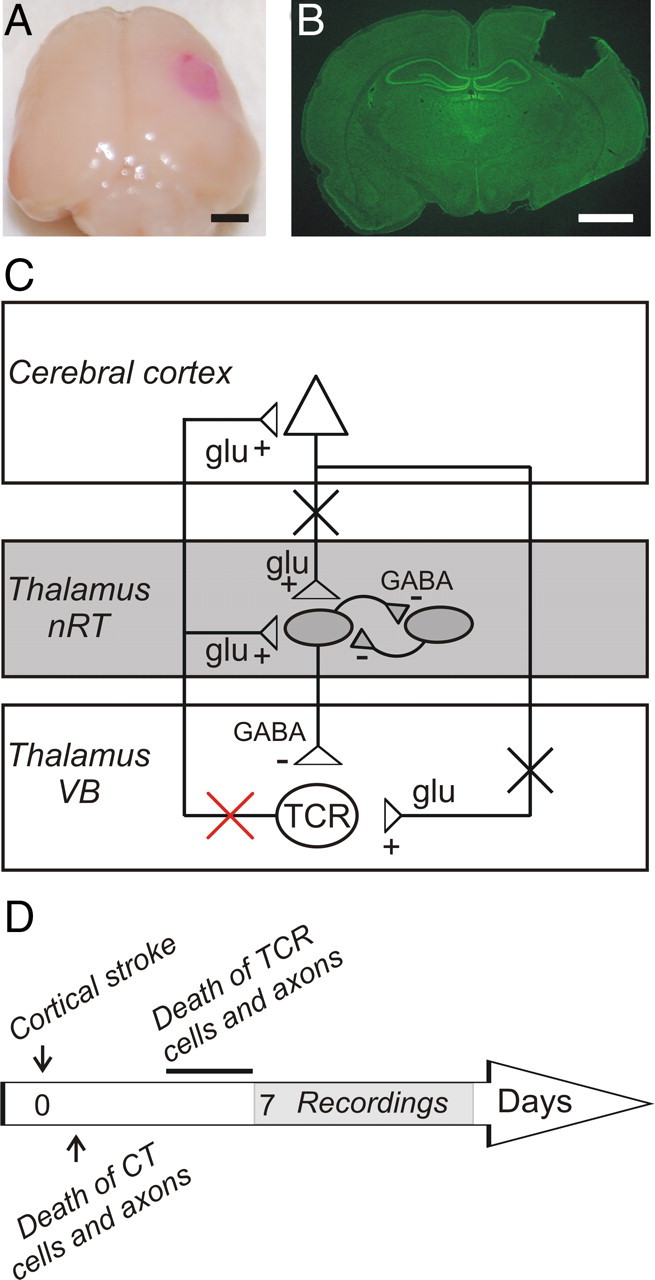
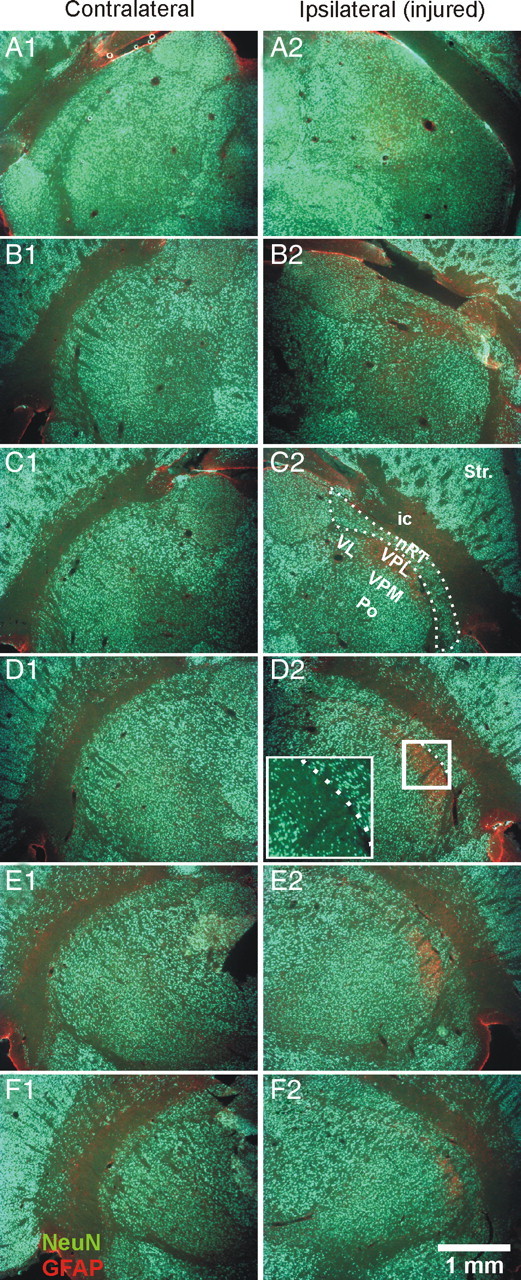
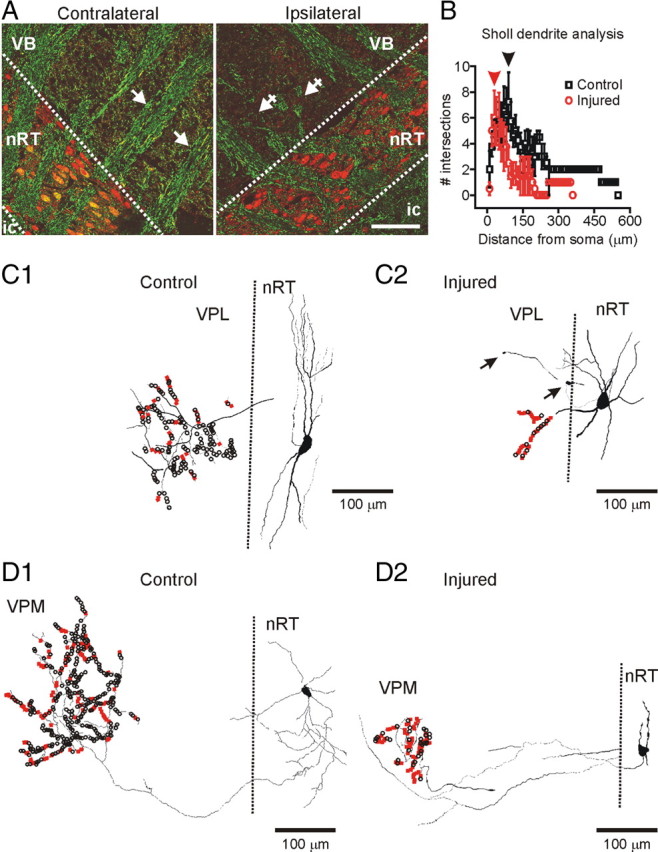
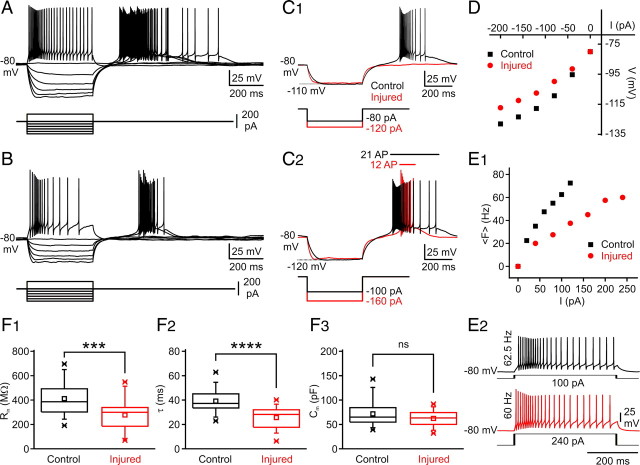
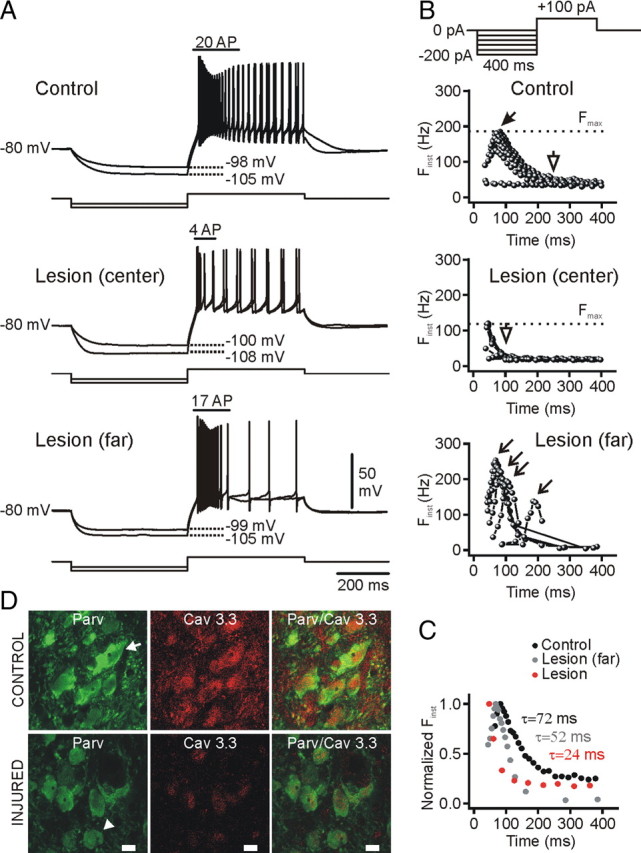
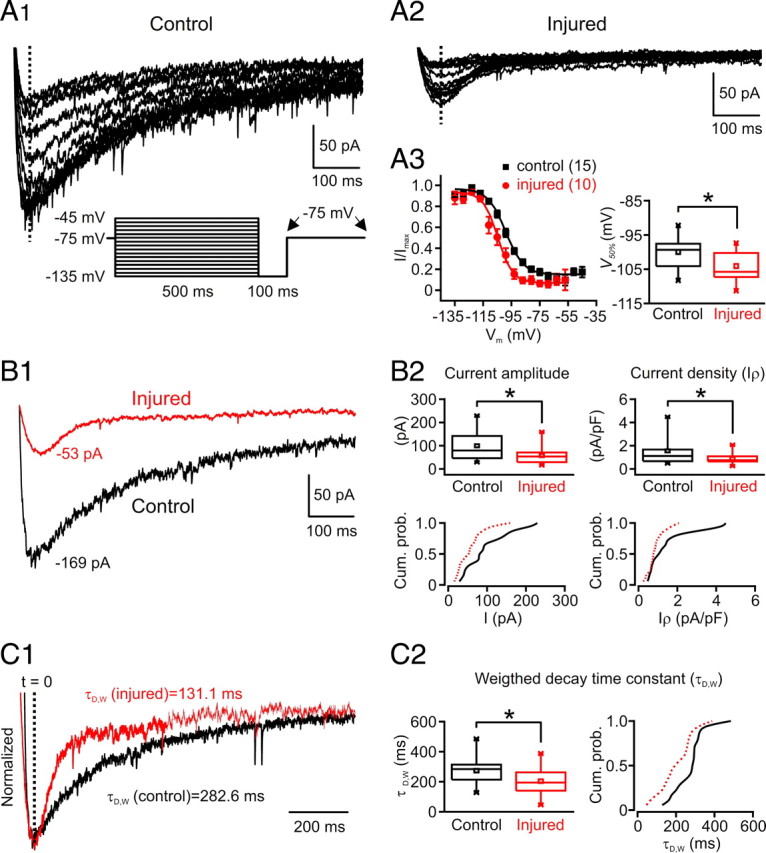
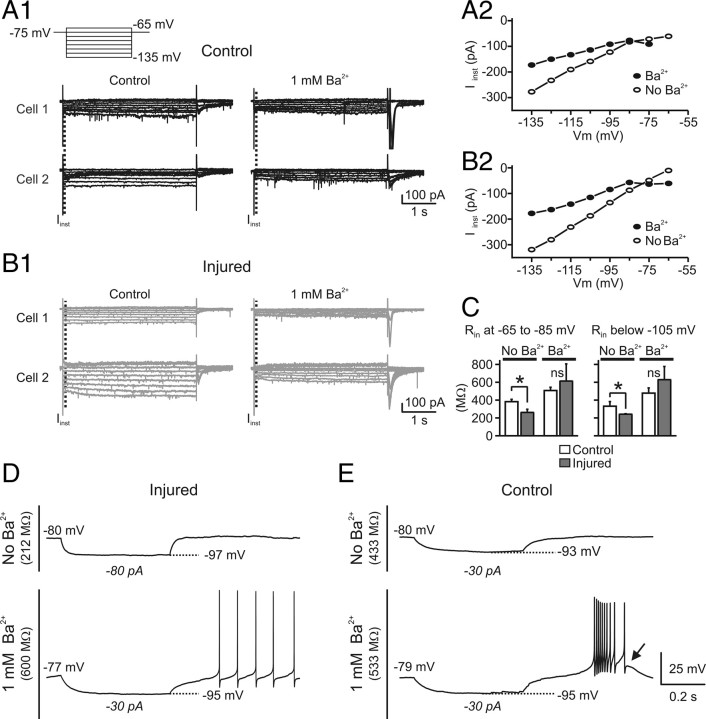
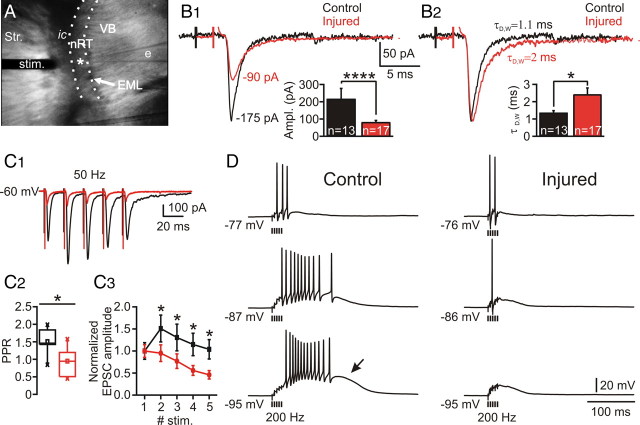
Focal cortical injuries result in death of cortical neurons and their efferents and ultimately in death or damage of thalamocortical relay (TCR) neurons that project to the affected cortical area. Neurons of the inhibitory reticular thalamic nucleus (nRT) receive excitatory inputs from corticothalamic and thalamocortical axons and are thus denervated by such injuries, yet nRT cells generally survive these insults to a greater degree than TCR cells. nRT cells inhibit TCR cells, regulate thalamocortical transmission, and generate cerebral rhythms including those involved in thalamocortical epilepsies. The survival and reorganization of nRT after cortical injury would determine recovery of thalamocortical circuits after injury. However, the physiological properties and connectivity of the survivors remain unknown. To study possible alterations in nRT neurons, we used the rat photothrombosis model of cortical stroke. Using in vitro patch-clamp recordings at various times after the photothrombotic injury, we show that localized strokes in the somatosensory cortex induce long-term reductions in intrinsic excitability and evoked synaptic excitation of nRT cells by the end of the first week after the injury. We find that nRT neurons in injured rats show (1) decreased membrane input resistance, (2) reduced low-threshold calcium burst responses, and (3) weaker evoked excitatory synaptic responses. Such alterations in nRT cellular excitability could lead to loss of nRT-mediated inhibition in relay nuclei, increased output of surviving TCR cells, and enhanced thalamocortical excitation, which may facilitate recovery of thalamic and cortical sensory circuits. In addition, such changes could be maladaptive, leading to injury-induced epilepsy.
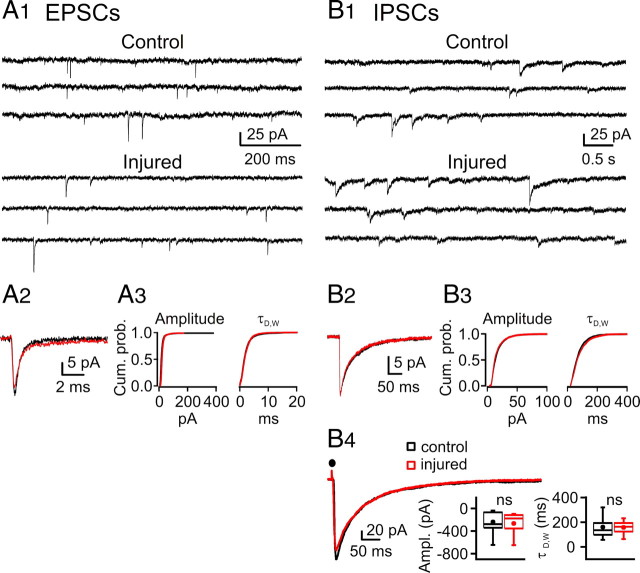
Figures
Similar articles
-
Two classes of excitatory synaptic responses in rat thalamic reticular neurons.J Neurophysiol. 2016 Sep 1;116(3):995-1011. doi: 10.1152/jn.01121.2015. Epub 2016 Jun 8. J Neurophysiol. 2016. PMID: 27281752 Free PMC article.
-
Physiology and pharmacology of corticothalamic stimulation-evoked responses in rat somatosensory thalamic neurons in vitro.J Neurophysiol. 1997 May;77(5):2661-76. doi: 10.1152/jn.1997.77.5.2661. J Neurophysiol. 1997. PMID: 9163382
-
Gamma-aminobutyric acid type B receptor-dependent burst-firing in thalamic neurons: a dynamic clamp study.Proc Natl Acad Sci U S A. 1996 Nov 12;93(23):13245-9. doi: 10.1073/pnas.93.23.13245. Proc Natl Acad Sci U S A. 1996. PMID: 8917576 Free PMC article.
-
Spike timing and synaptic dynamics at the awake thalamocortical synapse.Prog Brain Res. 2005;149:91-105. doi: 10.1016/S0079-6123(05)49008-1. Prog Brain Res. 2005. PMID: 16226579 Review.
-
The subplate and early cortical circuits.Annu Rev Neurosci. 2010;33:23-48. doi: 10.1146/annurev-neuro-060909-153244. Annu Rev Neurosci. 2010. PMID: 20201645 Review.
Cited by
-
Contribution of apoptosis-associated signaling pathways to epileptogenesis: lessons from Bcl-2 family knockouts.Front Cell Neurosci. 2013 Jul 16;7:110. doi: 10.3389/fncel.2013.00110. eCollection 2013. Front Cell Neurosci. 2013. PMID: 23882182 Free PMC article.
-
Weakened rate-dependent depression of Hoffmann's reflex and increased motoneuron hyperactivity after motor cortical infarction in mice.Cell Death Dis. 2014 Jan 16;5(1):e1007. doi: 10.1038/cddis.2013.544. Cell Death Dis. 2014. PMID: 24434515 Free PMC article.
-
The Ca(V)3.3 calcium channel is the major sleep spindle pacemaker in thalamus.Proc Natl Acad Sci U S A. 2011 Aug 16;108(33):13823-8. doi: 10.1073/pnas.1105115108. Epub 2011 Aug 1. Proc Natl Acad Sci U S A. 2011. PMID: 21808016 Free PMC article.
-
Morphological alterations of the reticular thalamic nucleus in Engrailed-2 knockout mice.J Anat. 2020 May;236(5):883-890. doi: 10.1111/joa.13150. Epub 2020 Jan 23. J Anat. 2020. PMID: 31972897 Free PMC article.
-
GABAA receptor in the thalamic specific relay system contributes to the propofol-induced somatosensory cortical suppression in rat.PLoS One. 2013 Dec 4;8(12):e82377. doi: 10.1371/journal.pone.0082377. eCollection 2013. PLoS One. 2013. PMID: 24324778 Free PMC article.
References
-
- Abbas SY, Ying SW, Goldstein PA. Compartmental distribution of hyperpolarization-activated cyclic-nucleotide-gated channel 2 and hyperpolarization-activated cyclic-nucleotide-gated channel 4 in thalamic reticular and thalamocortical relay neurons. Neuroscience. 2006;141:1811–1825. - PubMed
-
- Agarwala S, Kalil RE. Axotomy-induced neuronal death and reactive astrogliosis in the lateral geniculate nucleus following a lesion of the visual cortex in the rat. J Comp Neurol. 1998;392:252–263. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases