

Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology
- PMID: 20393462
- PMCID: PMC3796764
- DOI: 10.1038/nature08949
Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology
Abstract
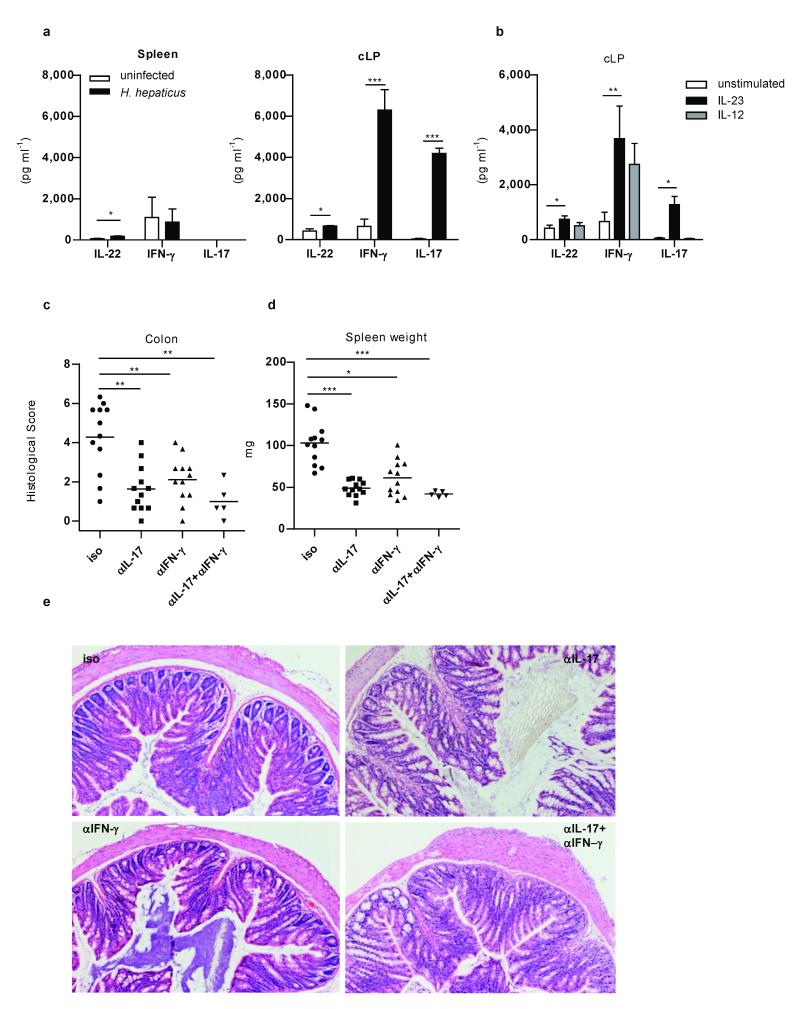
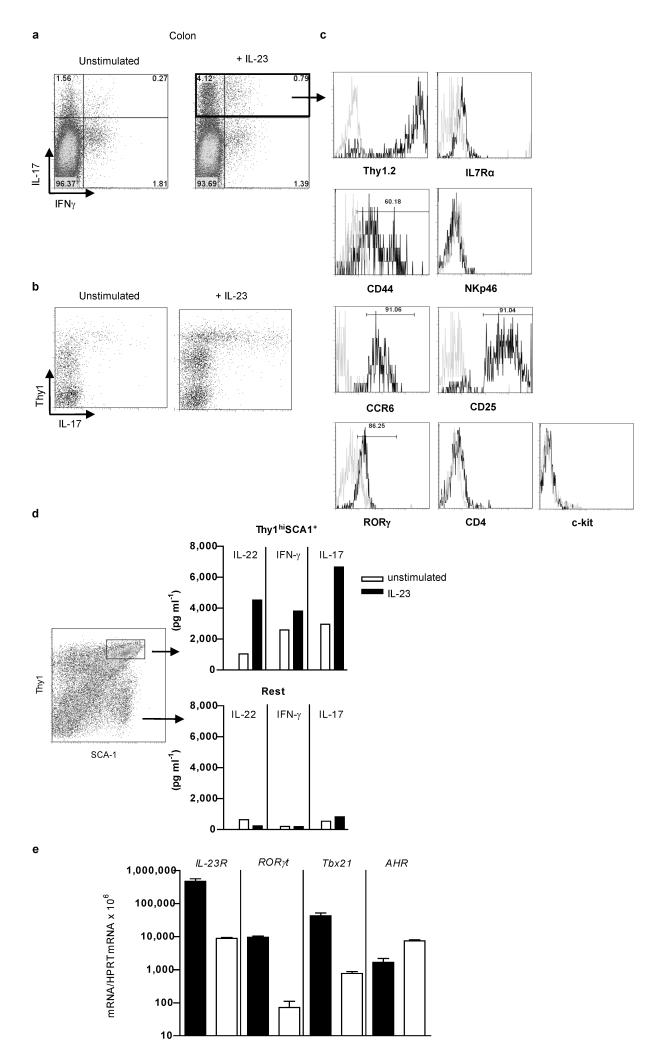
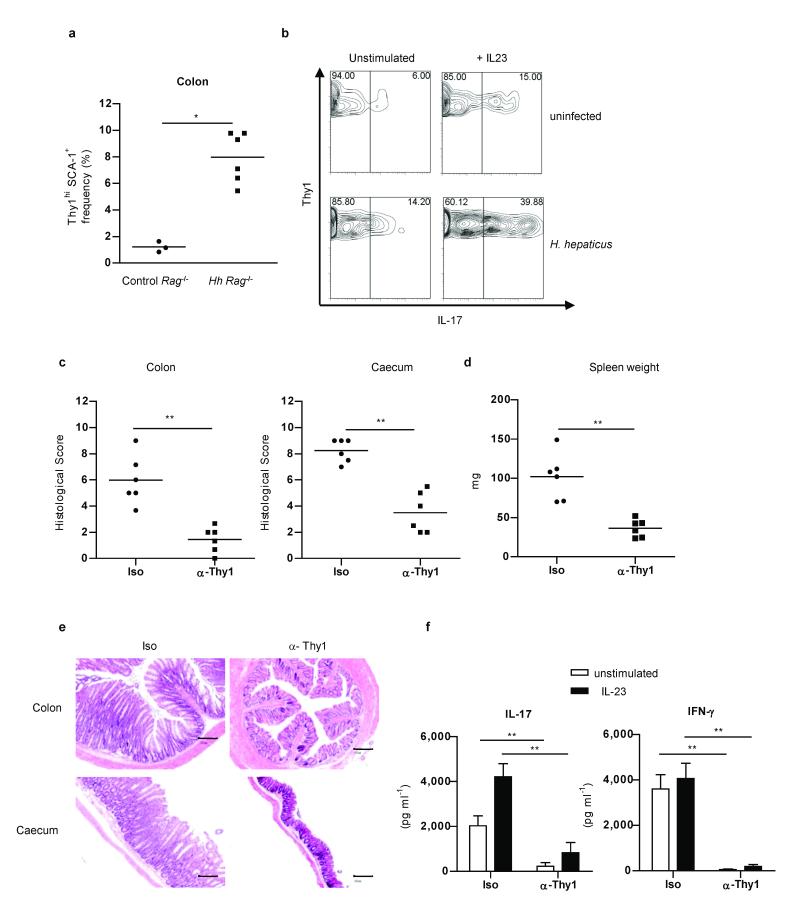
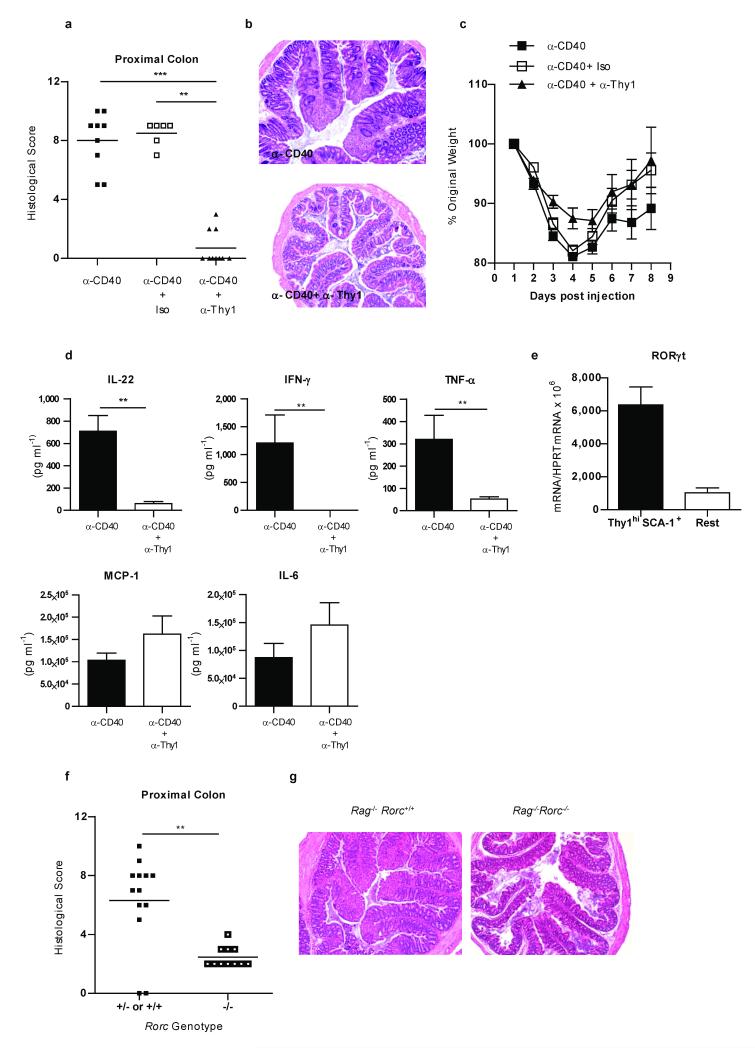
The key role of interleukin (IL)-23 in the pathogenesis of autoimmune and chronic inflammatory disorders is supported by the identification of IL-23 receptor (IL-23R) susceptibility alleles associated with inflammatory bowel disease, psoriasis and ankylosing spondylitis. IL-23-driven inflammation has primarily been linked to the actions of T-helper type 17 (TH17) cells. Somewhat overlooked, IL-23 also has inflammatory effects on innate immune cells and can drive T-cell-independent colitis. However, the downstream cellular and molecular pathways involved in this innate intestinal inflammatory response are poorly characterized. Here we show that bacteria-driven innate colitis is associated with an increased production of IL-17 and interferon-gamma in the colon. Stimulation of colonic leukocytes with IL-23 induced the production of IL-17 and interferon-gamma exclusively by innate lymphoid cells expressing Thy1, stem cell antigen 1 (SCA-1), retinoic-acid-related orphan receptor (ROR)-gammat and IL-23R, and these cells markedly accumulated in the inflamed colon. IL-23-responsive innate intestinal cells are also a feature of T-cell-dependent models of colitis. The transcription factor ROR-gammat, which controls IL-23R expression, has a functional role, because Rag-/-Rorc-/- mice failed to develop innate colitis. Last, depletion of Thy1+ innate lymphoid cells completely abrogated acute and chronic innate colitis. These results identify a previously unrecognized IL-23-responsive innate lymphoid population that mediates intestinal immune pathology and may therefore represent a target in inflammatory bowel disease.
Figures
References
-
- McGeachy MJ, Cua DJ. Th17 cell differentiation: the long and winding road. Immunity. 2008;28:445–453. - PubMed
-
- Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
