

Review
doi: 10.1038/nrg2774.
The primary cilium: a signalling centre during vertebrate development
Affiliations
- PMID: 20395968
- PMCID: PMC3121168
- DOI: 10.1038/nrg2774
Item in Clipboard
Review
The primary cilium: a signalling centre during vertebrate development
Nat Rev Genet.
2010 May.
Abstract
The primary cilium has recently stepped into the spotlight, as a flood of data show that this organelle has crucial roles in vertebrate development and human genetic diseases. Cilia are required for the response to developmental signals, and evidence is accumulating that the primary cilium is specialized for hedgehog signal transduction. The formation of cilia, in turn, is regulated by other signalling pathways, possibly including the planar cell polarity pathway. The cilium therefore represents a nexus for signalling pathways during development. The connections between cilia and developmental signalling have begun to clarify the basis of human diseases associated with ciliary dysfunction.
Figures
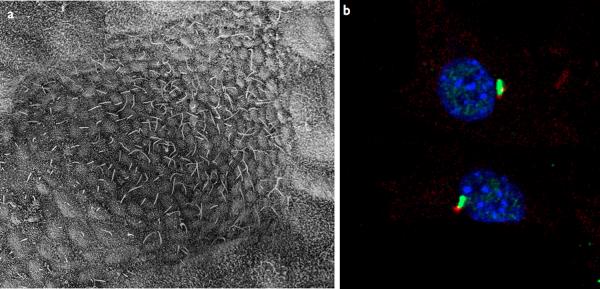
a–b | Examples of types of mammalian cilia. a | The long cilia of the node at E7.5 are required for left-right asymmetry. b | Primary cilia in embryonic fibroblasts in green with the basal body in red. c | Cargo is transported from the base to the tip of the cilium along the microtubule axoneme by Kinesin-2 together with the IFT-A and IFT-B complexes. Dynein mediates the return of IFT cargo to the base of the cilium, . IFT-B proteins IFT-20 and IFT-54 may also participate in the trafficking of membrane vesicles from the Golgi to the ciliary membrane together with small GTPases. Other small GTPases, including Arl13b also localize to cilia. While its precise trafficking role is not known, Arl13b is required for axoneme structure. Certain basal body proteins also influence ciliary trafficking. Among these are components of the Bbsome, named their association with Bardet-Biedl syndrome (BBS). The precise functions of BBS proteins in cilia formation are unclear as they are not individually required for primary cilia formation, however they may function to promote loading of cargo to the ciliary axoneme. Other basal body associated protein such as MKS1, FTM, OFD1, and Talpid3 are required for cilia formation, though how they regulate ciliogenesis has not been defined (For a review, see).
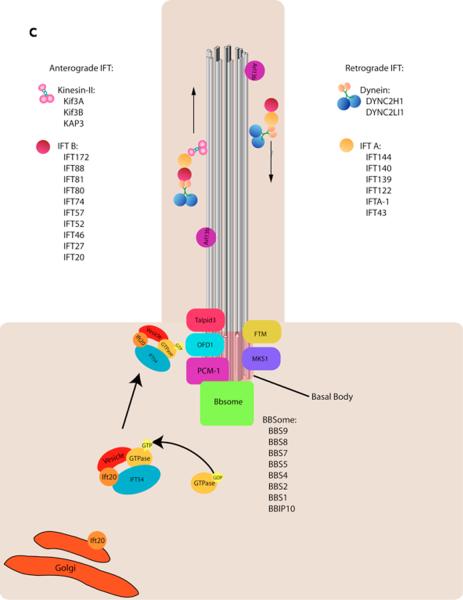
a–b | Examples of types of mammalian cilia. a | The long cilia of the node at E7.5 are required for left-right asymmetry. b | Primary cilia in embryonic fibroblasts in green with the basal body in red. c | Cargo is transported from the base to the tip of the cilium along the microtubule axoneme by Kinesin-2 together with the IFT-A and IFT-B complexes. Dynein mediates the return of IFT cargo to the base of the cilium, . IFT-B proteins IFT-20 and IFT-54 may also participate in the trafficking of membrane vesicles from the Golgi to the ciliary membrane together with small GTPases. Other small GTPases, including Arl13b also localize to cilia. While its precise trafficking role is not known, Arl13b is required for axoneme structure. Certain basal body proteins also influence ciliary trafficking. Among these are components of the Bbsome, named their association with Bardet-Biedl syndrome (BBS). The precise functions of BBS proteins in cilia formation are unclear as they are not individually required for primary cilia formation, however they may function to promote loading of cargo to the ciliary axoneme. Other basal body associated protein such as MKS1, FTM, OFD1, and Talpid3 are required for cilia formation, though how they regulate ciliogenesis has not been defined (For a review, see).
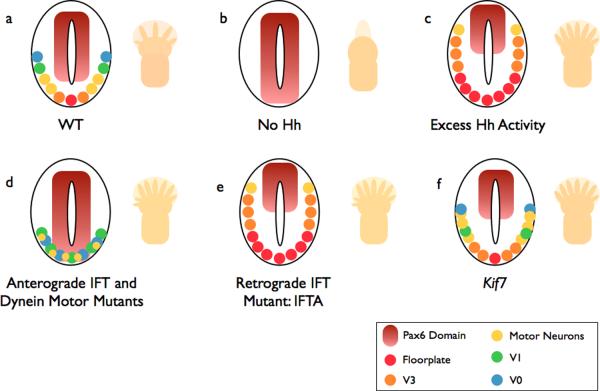
a | In wild-type (WT) embryos, ventral neural cell fates are specified by a gradient of Shh. The number and identity of digits in the limb is established by a Shh gradient from the posterior limb bud opposed by anterior Gli3 repressor (Gli3R). b | In the absence of Hh, (e.g. Smo −/−), ventral neural cell fates are lost. The limbs of Shh mutants lack digits. c | If the pathway is hyperactive (e.g. Ptch −/−), ventral cell types expand in the neural tube. Activation of the pathway within the limb, such as in Gli3 mutants (which lack Gli3R), causes the formation of extra digits. d | Anterograde IFT mutants (e.g. the IFT-B mutants Ift88 or Ift172) lack cilia: Hh signaling is reduced, and the neural tube is dorsalized. This phenotype is milder than in (b) because cilia are also required for cilia Gli3R processing, and cell types that require low levels of Hh signaling are specified. Reduced Gli3R results in polydactyly. Dynein mutants display a similar phenotype, however they retain motor neurons in the caudal neural tube. e | IFT-A mutants (ie Ift139) exhibit phenotypes consistent with excess Hh signaling. f | Mutations that disrupt the kinesin Kif7 cause a partial activation of the Hh pathway, with a modest expansion of cells that require intermediate levels of Hh.
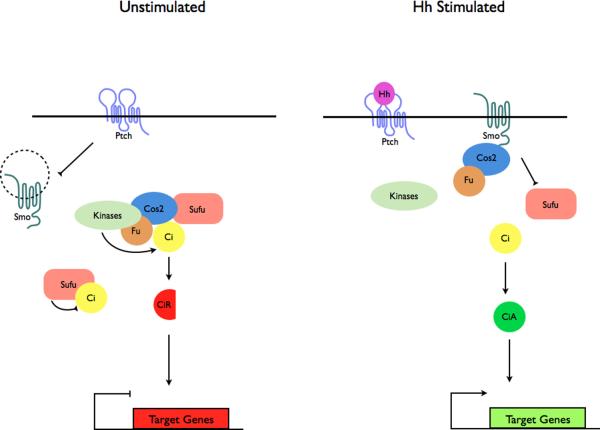
a | Modulation of protein complex structure and localization in Drosophila by Hh. In the absence of ligand, Ptch prevents translocation of Smo to the plasma membrane. A microtubule-associated complex including Cos2, Fu, Sufu and Ci recruits kinases including PKA, CK1, GSK3β that promote the processing of Ci into its repressor form (CiR)–. Sufu may also associate with full length Ci to prevent its translocation to the nucleus. Upon activation of the pathway, Smo moves to the plasma membrane, and the Fu-Cos2 complex associates with the C-terminal tail of Smo resulting in the release of Ci. Pathway activation also inactivates the negative regulator–. b | In vertebrate Hh signaling, signal transduction takes place within cilia, but the behavior of protein complexes may parallel that in Drosophila. In the absence of ligand, Ptch localizes to the cilium, and is thought to block the entry of Smo into cilia. Kif7 (the Cos2 homologue) localizes to the base of the cilium where it may form a complex Gli proteins and other pathway components. Kif7 at the cilium base prevents Gli enrichment within the cilium and promotes processing of GliR. Upon activation of the pathway, Smo moves to the ciliary membrane and Kif7 translocates into the cilium thereby promoting Gli2 accumulation at the cilium tip, . Kif7 at the cilia tip may also block the function of Sufu. Activated Gli is transported out of the cilium by Dynein and IFT particles.
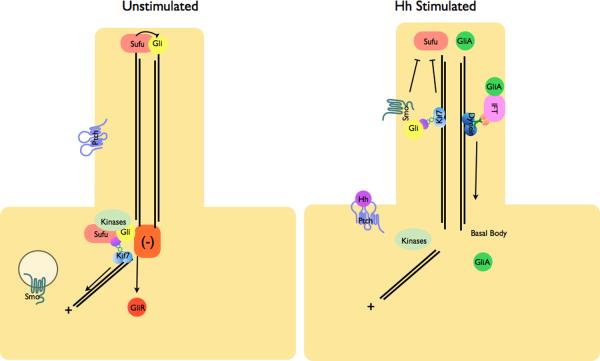
a | Modulation of protein complex structure and localization in Drosophila by Hh. In the absence of ligand, Ptch prevents translocation of Smo to the plasma membrane. A microtubule-associated complex including Cos2, Fu, Sufu and Ci recruits kinases including PKA, CK1, GSK3β that promote the processing of Ci into its repressor form (CiR)–. Sufu may also associate with full length Ci to prevent its translocation to the nucleus. Upon activation of the pathway, Smo moves to the plasma membrane, and the Fu-Cos2 complex associates with the C-terminal tail of Smo resulting in the release of Ci. Pathway activation also inactivates the negative regulator–. b | In vertebrate Hh signaling, signal transduction takes place within cilia, but the behavior of protein complexes may parallel that in Drosophila. In the absence of ligand, Ptch localizes to the cilium, and is thought to block the entry of Smo into cilia. Kif7 (the Cos2 homologue) localizes to the base of the cilium where it may form a complex Gli proteins and other pathway components. Kif7 at the cilium base prevents Gli enrichment within the cilium and promotes processing of GliR. Upon activation of the pathway, Smo moves to the ciliary membrane and Kif7 translocates into the cilium thereby promoting Gli2 accumulation at the cilium tip, . Kif7 at the cilia tip may also block the function of Sufu. Activated Gli is transported out of the cilium by Dynein and IFT particles.
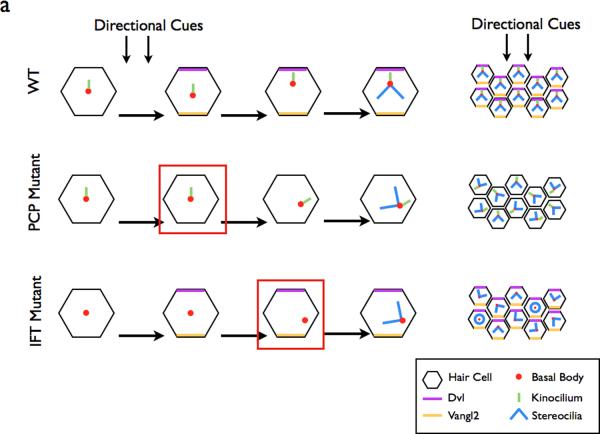
a | In the WT cochlea, directional cues establish localization of core PCP pathway components. The kinocilium then directs the basal body towards the medial side of the cells. This in turn directs the orientation of the stereocilia bundles, resulting in the correct orientation of the bundles within the cochlea. In PCP mutants, the initial cell polarity is never established, resulting in the improper positioning of the basal body and stereocilia. In IFT mutants, planar polarity is established, however the basal body is not repositioned in the absence of the IFT-dependent kinocilium. Red boxes depict the step in the establishment of ppolarity that is defective in each category of mutants. b | Components of the PCP pathway are implicated in cilia formation. The centrioles are initially located away from the cell surface (1) where they associate with Dvl, a PCP protein. Sec8, a component of vesicle trafficking machinery is recruited to the basal body in (2). At the cell surface in (3), Dvl mediates the fusion of the basal body-associated membrane with the cell membrane, . The axoneme of the cilium extends in (4). In vivo mouse experiments indicate that the PCP effector Fuzzy, together with the small GTPase RGS1, also plays a role in trafficking membrane vesicles and ciliary components to the basal body.
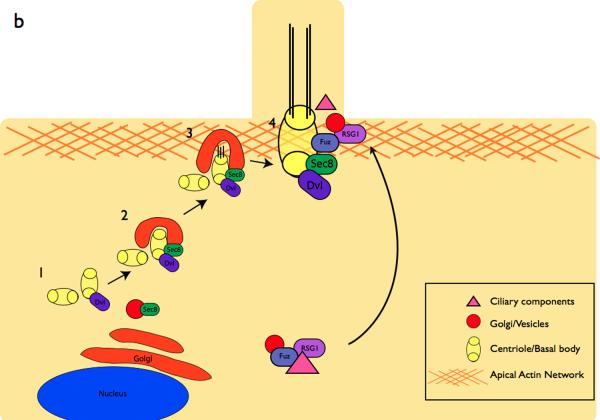
a | In the WT cochlea, directional cues establish localization of core PCP pathway components. The kinocilium then directs the basal body towards the medial side of the cells. This in turn directs the orientation of the stereocilia bundles, resulting in the correct orientation of the bundles within the cochlea. In PCP mutants, the initial cell polarity is never established, resulting in the improper positioning of the basal body and stereocilia. In IFT mutants, planar polarity is established, however the basal body is not repositioned in the absence of the IFT-dependent kinocilium. Red boxes depict the step in the establishment of ppolarity that is defective in each category of mutants. b | Components of the PCP pathway are implicated in cilia formation. The centrioles are initially located away from the cell surface (1) where they associate with Dvl, a PCP protein. Sec8, a component of vesicle trafficking machinery is recruited to the basal body in (2). At the cell surface in (3), Dvl mediates the fusion of the basal body-associated membrane with the cell membrane, . The axoneme of the cilium extends in (4). In vivo mouse experiments indicate that the PCP effector Fuzzy, together with the small GTPase RGS1, also plays a role in trafficking membrane vesicles and ciliary components to the basal body.
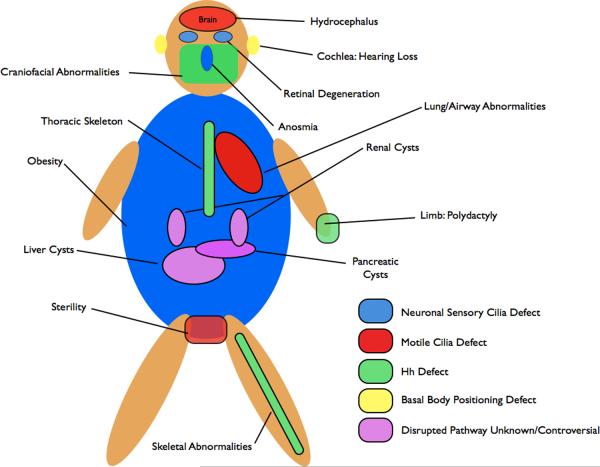
Numerous pleiotropic human disorders have been attributed to defects in cilia formation, . Some aspects of these syndromes, such as the polydactyly in patients with Bardet-Biedl syndrome (BBS) and Meckel Syndrome (MKS) and the skeletal abnormalities affecting the limbs in Ellis-van Creveld are attributed to defective Hh signaling. Polydactyly results from a loss of Gli3R and skeletal abnormalities resemble those observed in mutants that lack Ihh signaling. Shh signaling is also required for craniofacial development, and defects in craniofacial structures, such as those observed in Mks1 mutant mice, are also likely due to mis-regulated Hh signaling. In addition, patients with a Joubert syndrome-like disorder exhibit ataxia due to cerebellar hypoplasia. Growth of this tissue is Hh dependent. Other attributes of human disorders result from defective specialized cilia. Retinal degeneration results from defects to photoreceptor cilia, which connect the outer light-responsive segment to the cell body. Detection of odorants depends on the primary cilia of sensory neurons in the olfactory epithelium and BBS patients often exhibit anosmia–. Another sensory deficit, hearing loss, is due to a requirement for the specialized primary cilia of the cochlea downstream of the PCP pathway in establishing the correct polarity of sensory hair cells. Infertility observed in patients with ciliopathies is the result of defective motile cilia of spermatids or oviducts. For some of the most severe and common abnormalities associated with ciliopathies, such as cyst formation in the kidneys, liver, biliary duct and pancreas, the underlying molecular causes downstream of the cilium remain unclear. Cyst formation is thought to result from defects in cell proliferation or mis-orientation of the mitotic spindle, however, whether and how these processes are regulated by cilia remain the subject of active investigation. In addition, BBS patients often exhibit obesity and cognative impairments thought to be due to neuronal defects, however the specific pathways responsible for these attributes in BBS patients have not been clearly identified, .
References
-
- Pedersen LB, Veland IR, Schroder JM, Christensen ST. Assembly of primary cilia. Dev Dyn. 2008;237:1993–2006. - PubMed
-
- Silverman MA, Leroux MR. Intraflagellar transport and the generation of dynamic, structurally and functionally diverse cilia. Trends Cell Biol. 2009;19:306–16. - PubMed
-
- Zhou J. Polycystins and primary cilia: primers for cell cycle progression. Annu Rev Physiol. 2009;71:83–113. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
