

Varicella-zoster virus T cell tropism and the pathogenesis of skin infection
- PMID: 20397071
- PMCID: PMC4077053
- DOI: 10.1007/82_2010_29
Varicella-zoster virus T cell tropism and the pathogenesis of skin infection
Abstract
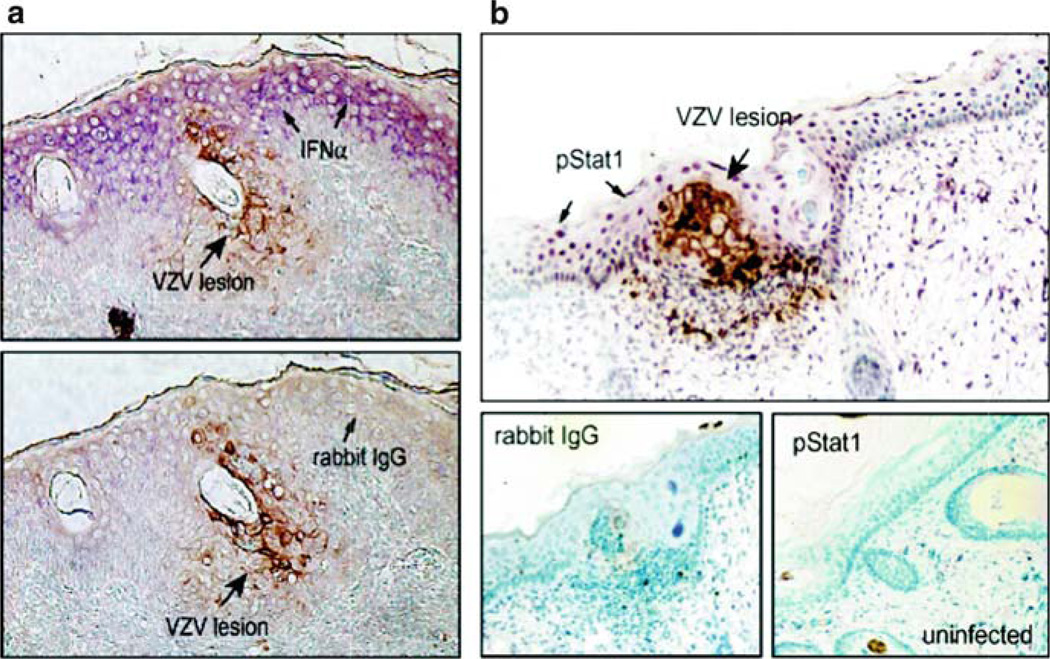
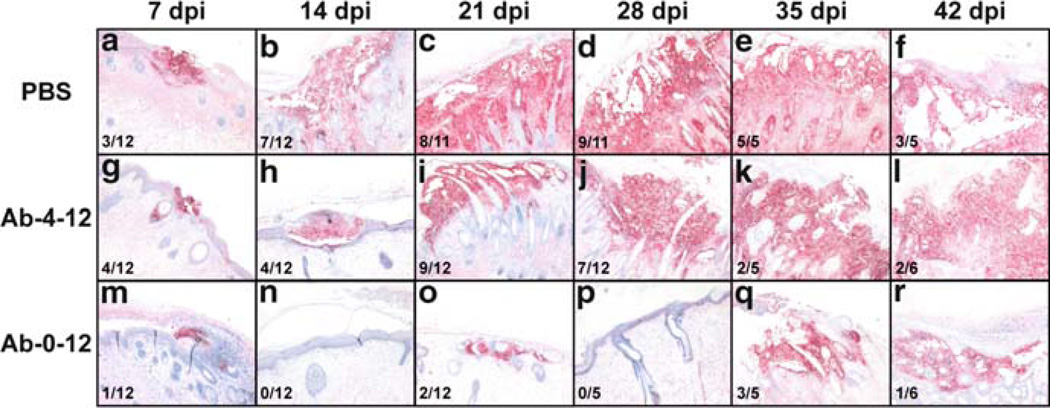
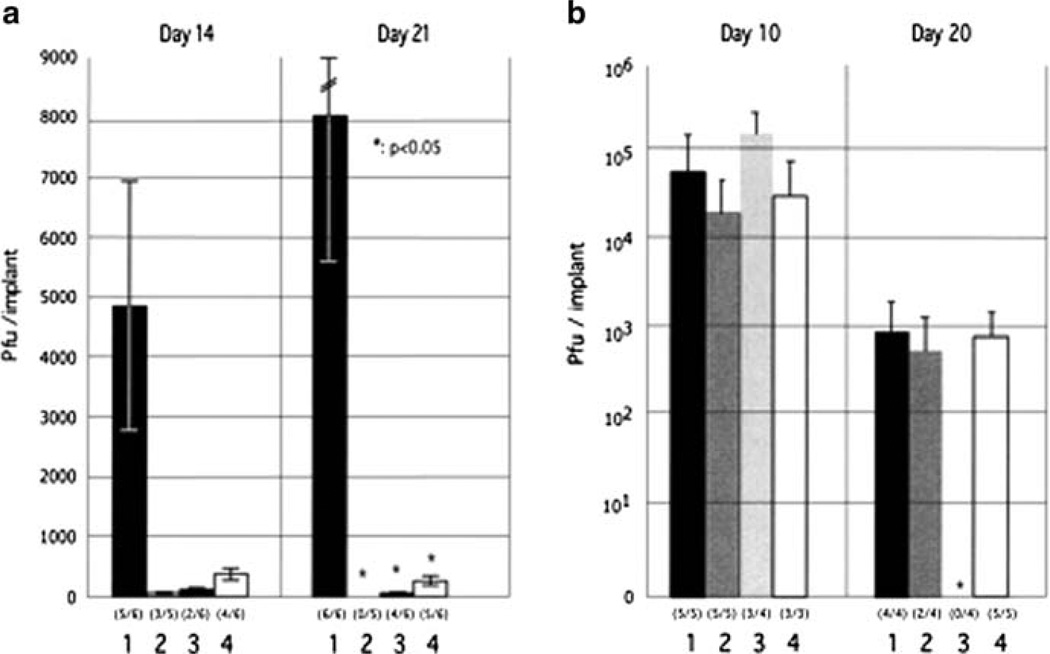
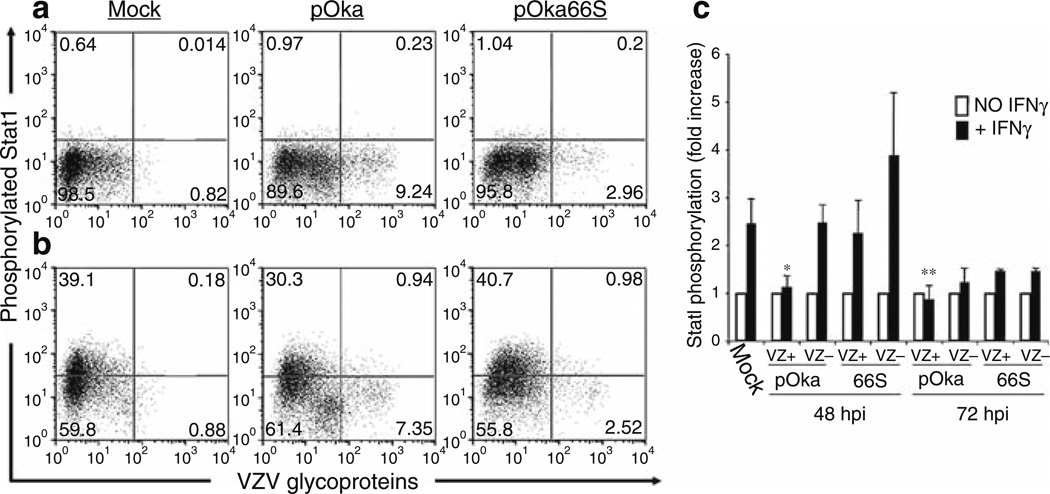
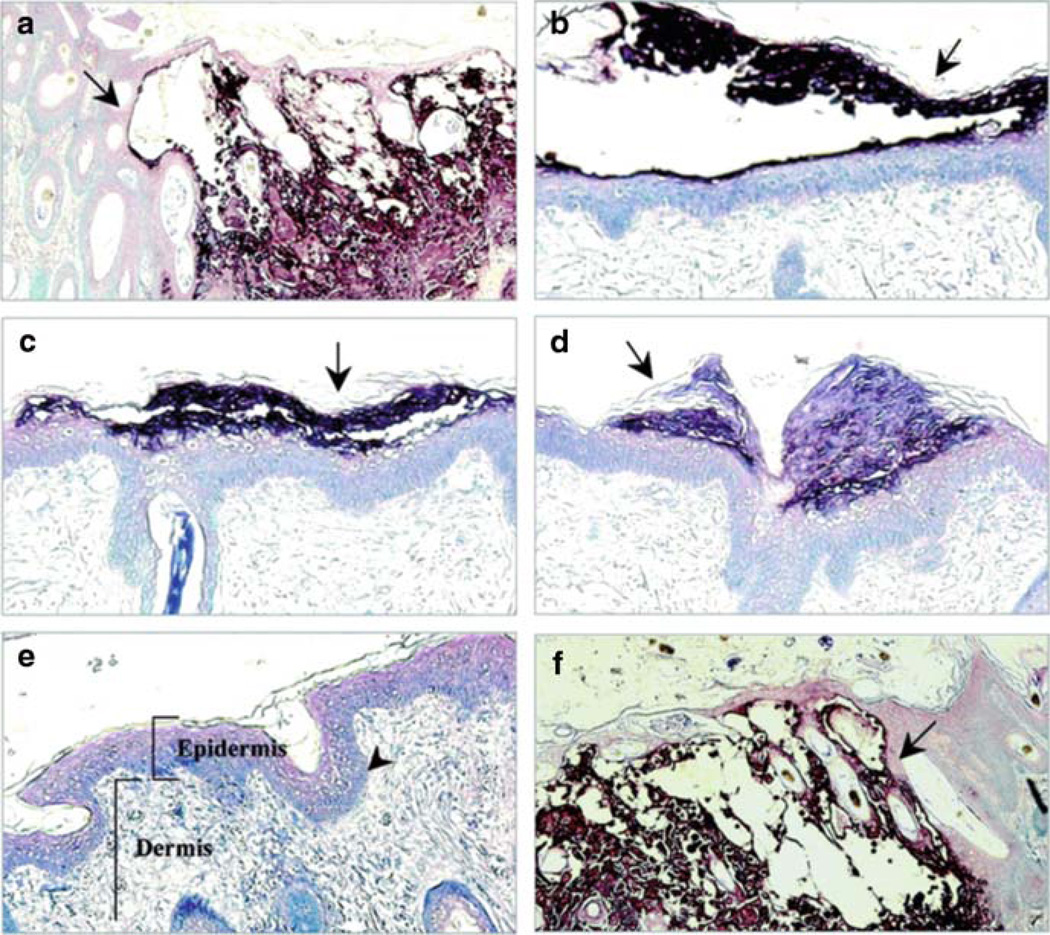
Varicella-zoster virus (VZV) is a medically important human alphaherpesvirus that causes varicella and zoster. VZV initiates primary infection by inoculation of the respiratory mucosa. In the course of primary infection, VZV establishes a life-long persistence in sensory ganglia; VZV reactivation from latency may result in zoster in healthy and immunocompromised patients. The VZV genome has at least 70 known or predicted open reading frames (ORFs), but understanding how these gene products function in virulence is difficult because VZV is a highly human-specific pathogen. We have addressed this obstacle by investigating VZV infection of human tissue xenografts in the severe combined immunodeficiency mouse model. In studies relevant to the pathogenesis of primary VZV infection, we have examined VZV infection of human T cell (thymus/liver) and skin xenografts. This work supports a new paradigm for VZV pathogenesis in which VZV T cell tropism provides a mechanism for delivering the virus to skin. We have also shown that VZV-infected T cells transfer VZV to neurons in sensory ganglia. The construction of infectious VZV recombinants that have deletions or targeted mutations of viral genes or their promoters and the evaluation of VZV mutants in T cell and skin xenografts has revealed determinants of VZV virulence that are important for T cell and skin tropism in vivo.
Figures
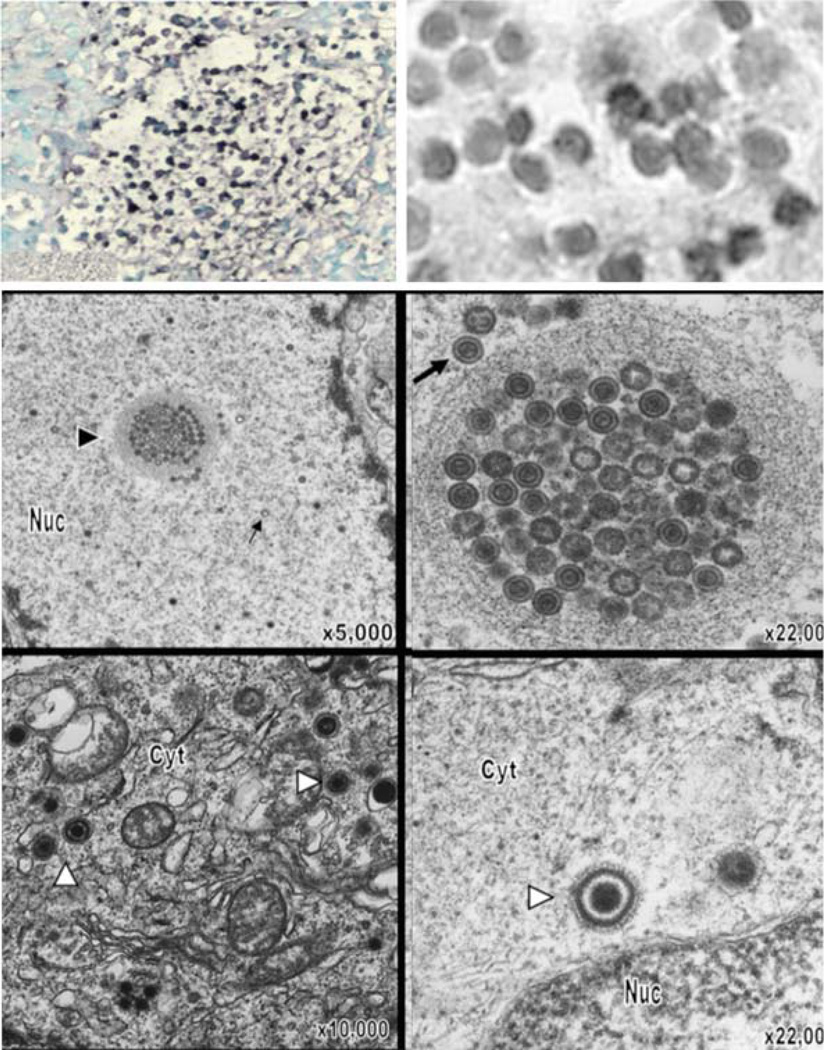
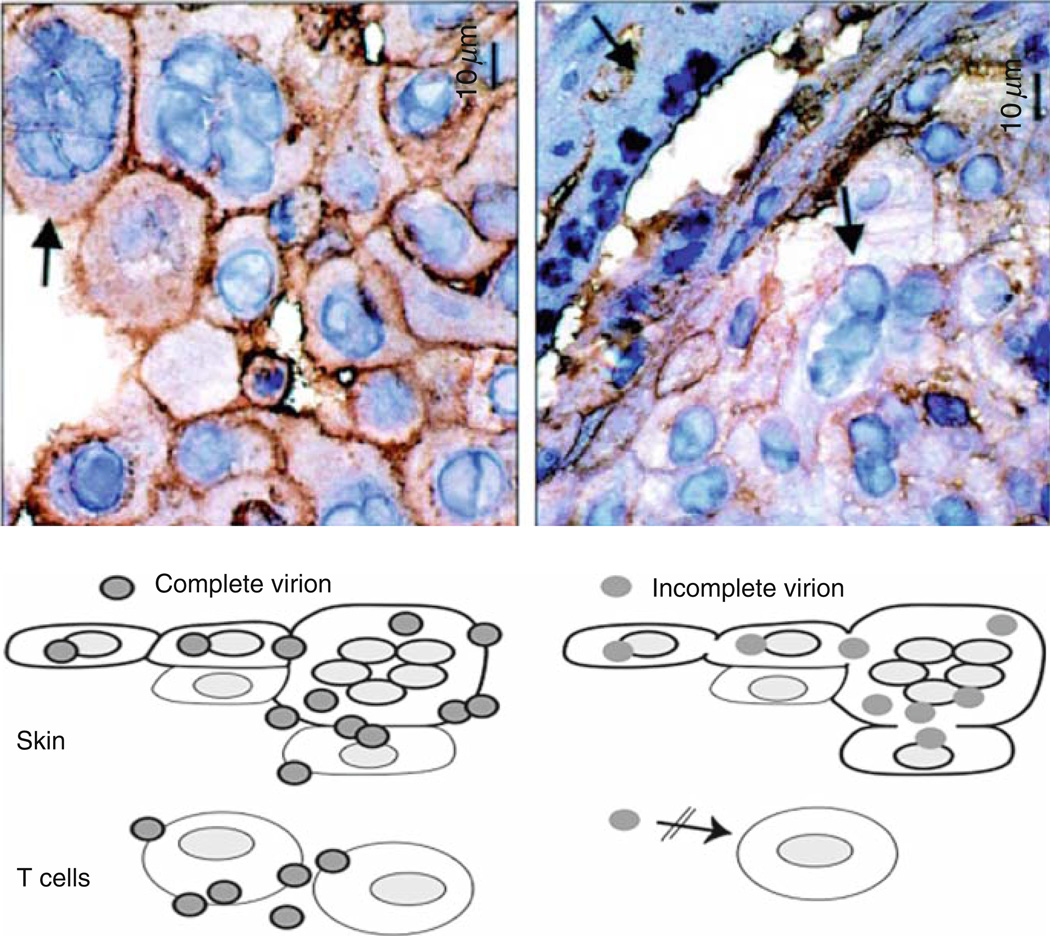
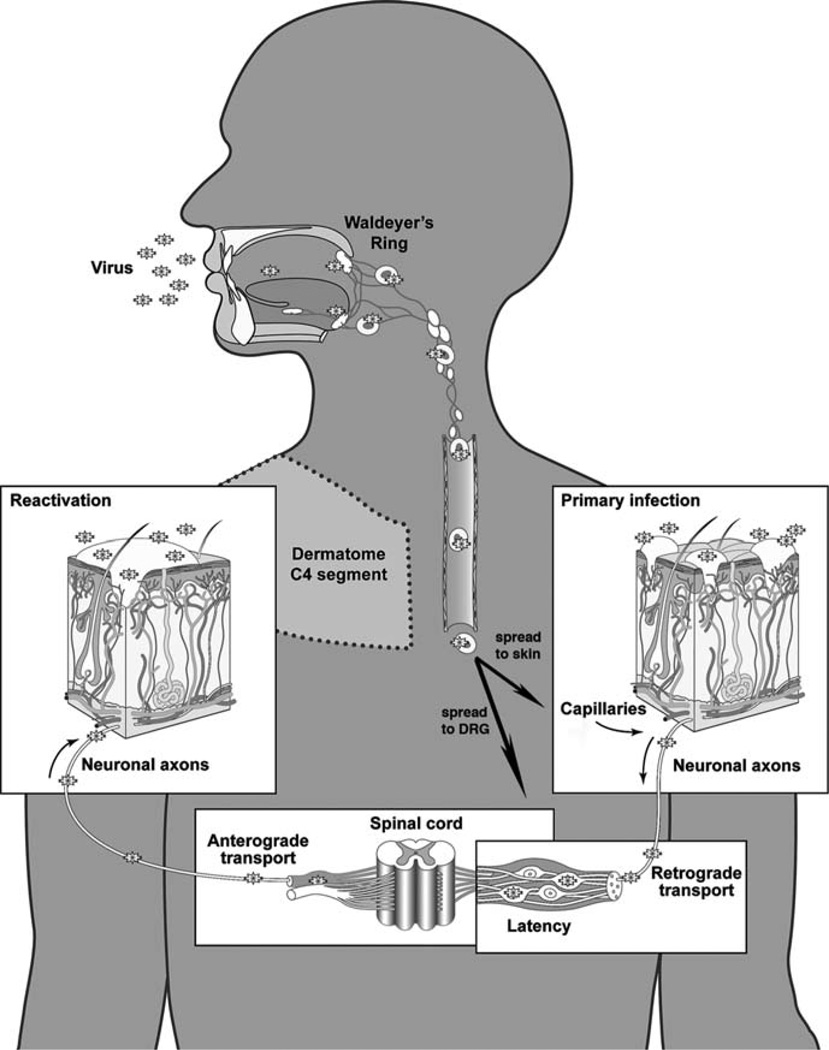
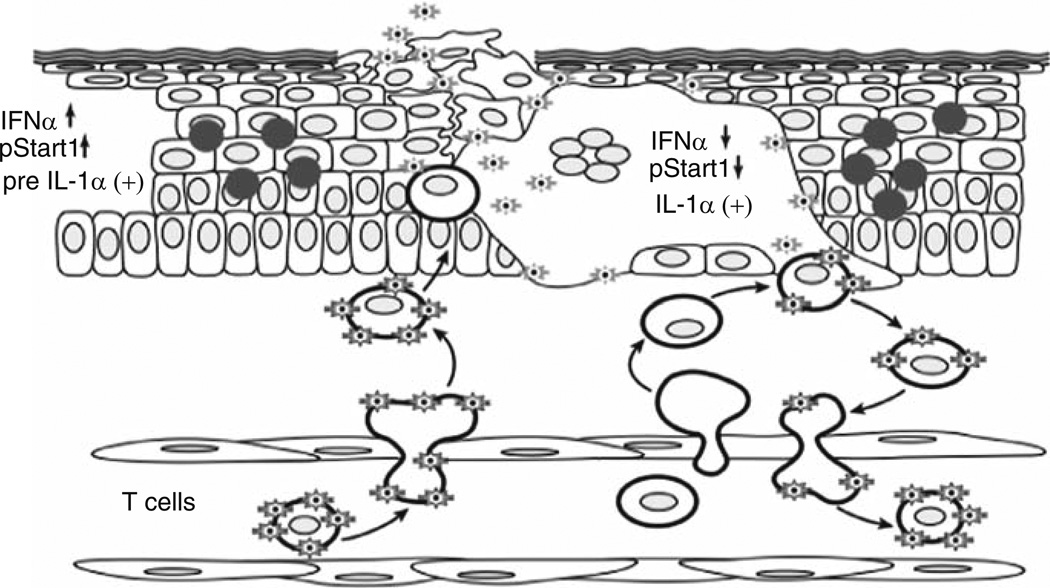
References
-
- Arvin AM. Varicella-zoster virus. In: Knipe DM, Howley P, editors. Fields’ virology. 4th edn. Philadelphia: Lippincott-Williams & Wilkins; 2001a. pp. 2731–2768.
-
- Arvin AM. Varicella vaccine: genesis, attenuation and efficacy. Virology. 2001b;284:153–158. - PubMed
-
- Arvin AM, Schaap AC, Ku C-C, Jones JO, Sommer M, Zerboni Z. Investigations of the molecular mechanisms of varicella-zoster virus pathogenesis. In: Sandri-Golden R, editor. The alphaherpesviruses. Horizon Press Inc: UK: 2006.
-
- Baiker A, Bagowski C, Ito H, Sommer M, Zerboni L, Fabell K, Hay J, Ruyechan W, Arvin AM. The immediate early 63 protein of varicella-zoster virus: analysis of functional domains required for replication in vitro and for T cell and skin tropism in the SCID hu model in vivo. J Virol. 2004;78:1181–1194. - PMC - PubMed
-
- Besser J, Sommer MH, Zerboni L, Bagowski C, Ito H, Moffat J, Ku C-C, Arvin AM. Differentiation of varicella-zoster virus ORF47 protein kinase and IE62 protein binding domains and their contributions to replication in human skin xenografts in the SCID-hu mouse. J Virol. 2003;77:5964–5974. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
