

Orientation-dependent backbone-only residue pair scoring functions for fixed backbone protein design
- PMID: 20398384
- PMCID: PMC2874805
- DOI: 10.1186/1471-2105-11-192
Orientation-dependent backbone-only residue pair scoring functions for fixed backbone protein design
Abstract
Background: Empirical scoring functions have proven useful in protein structure modeling. Most such scoring functions depend on protein side chain conformations. However, backbone-only scoring functions do not require computationally intensive structure optimization and so are well suited to protein design, which requires fast score evaluation. Furthermore, scoring functions that account for the distinctive relative position and orientation preferences of residue pairs are expected to be more accurate than those that depend only on the separation distance.
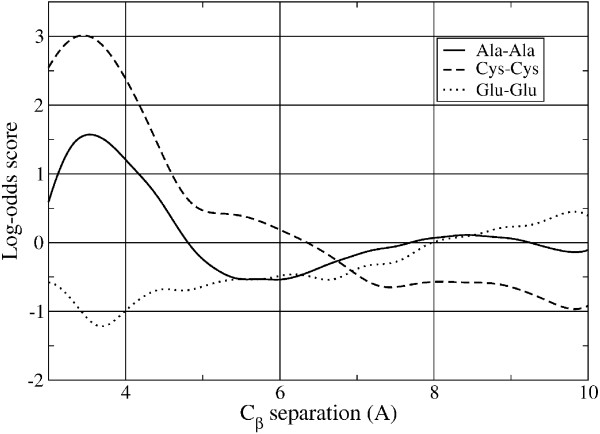
Results: Residue pair scoring functions for fixed backbone protein design were derived using only backbone geometry. Unlike previous studies that used spherical harmonics to fit 2D angular distributions, Gaussian Mixture Models were used to fit the full 3D (position only) and 6D (position and orientation) distributions of residue pairs. The performance of the 1D (residue separation only), 3D, and 6D scoring functions were compared by their ability to identify correct threading solutions for a non-redundant benchmark set of protein backbone structures. The threading accuracy was found to steadily increase with increasing dimension, with the 6D scoring function achieving the highest accuracy. Furthermore, the 3D and 6D scoring functions were shown to outperform side chain-dependent empirical potentials from three other studies. Next, two computational methods that take advantage of the speed and pairwise form of these new backbone-only scoring functions were investigated. The first is a procedure that exploits available sequence data by averaging scores over threading solutions for homologs. This was evaluated by applying it to the challenging problem of identifying interacting transmembrane alpha-helices and found to further improve prediction accuracy. The second is a protein design method for determining the optimal sequence for a backbone structure by applying Belief Propagation optimization using the 6D scoring functions. The sensitivity of this method to backbone structure perturbations was compared with that of fixed-backbone all-atom modeling by determining the similarities between optimal sequences for two different backbone structures within the same protein family. The results showed that the design method using 6D scoring functions was more robust to small variations in backbone structure than the all-atom design method.
Conclusions: Backbone-only residue pair scoring functions that account for all six relative degrees of freedom are the most accurate and including the scores of homologs further improves the accuracy in threading applications. The 6D scoring function outperformed several side chain-dependent potentials while avoiding time-consuming and error prone side chain structure prediction. These scoring functions are particularly useful as an initial filter in protein design problems before applying all-atom modeling.
Figures
Similar articles
-
KORP: knowledge-based 6D potential for fast protein and loop modeling.Bioinformatics. 2019 Sep 1;35(17):3013-3019. doi: 10.1093/bioinformatics/btz026. Bioinformatics. 2019. PMID: 30649193
-
A consistent set of statistical potentials for quantifying local side-chain and backbone interactions.Proteins. 2005 Jul 1;60(1):90-6. doi: 10.1002/prot.20482. Proteins. 2005. PMID: 15852305
-
A pair-conformation-dependent scoring function for evaluating 3D RNA-protein complex structures.PLoS One. 2017 Mar 30;12(3):e0174662. doi: 10.1371/journal.pone.0174662. eCollection 2017. PLoS One. 2017. PMID: 28358834 Free PMC article.
-
From local structure to a global framework: recognition of protein folds.J R Soc Interface. 2014 Apr 16;11(95):20131147. doi: 10.1098/rsif.2013.1147. Print 2014 Jun 6. J R Soc Interface. 2014. PMID: 24740960 Free PMC article. Review.
-
Recent advances in de novo protein design: Principles, methods, and applications.J Biol Chem. 2021 Jan-Jun;296:100558. doi: 10.1016/j.jbc.2021.100558. Epub 2021 Mar 18. J Biol Chem. 2021. PMID: 33744284 Free PMC article. Review.
Cited by
-
ROTAS: a rotamer-dependent, atomic statistical potential for assessment and prediction of protein structures.BMC Bioinformatics. 2014 Sep 18;15(1):307. doi: 10.1186/1471-2105-15-307. BMC Bioinformatics. 2014. PMID: 25236673 Free PMC article.
References
-
- Miyazawa S, Jernigan RL. Estimation of effective interresidue contact energies from protein crystal structures: Quasi-chemical approximation. Macromolecules. 1985;18(3):534–552. doi: 10.1021/ma00145a039. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
