

A genome-wide survey of sRNAs in the symbiotic nitrogen-fixing alpha-proteobacterium Sinorhizobium meliloti
- PMID: 20398411
- PMCID: PMC2873474
- DOI: 10.1186/1471-2164-11-245
A genome-wide survey of sRNAs in the symbiotic nitrogen-fixing alpha-proteobacterium Sinorhizobium meliloti
Abstract
Background: Small untranslated RNAs (sRNAs) are widespread regulators of gene expression in bacteria. This study reports on a comprehensive screen for sRNAs in the symbiotic nitrogen-fixing alpha-proteobacterium Sinorhizobium meliloti applying deep sequencing of cDNAs and microarray hybridizations.
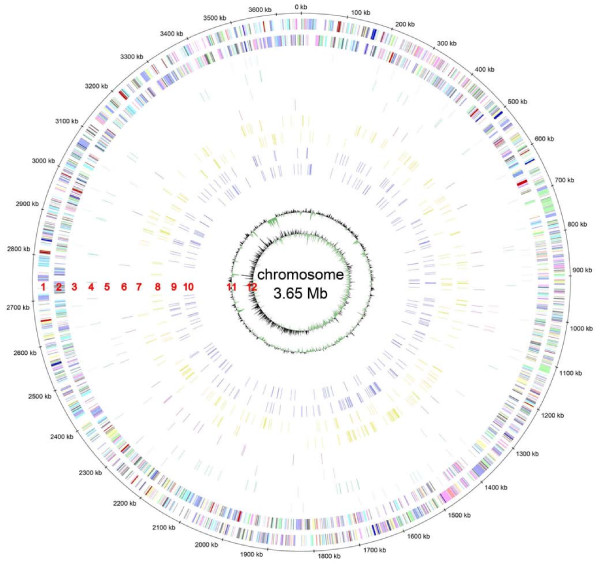
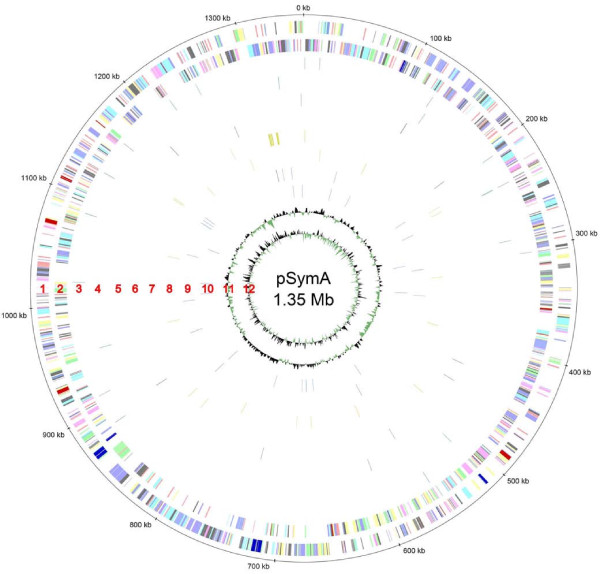
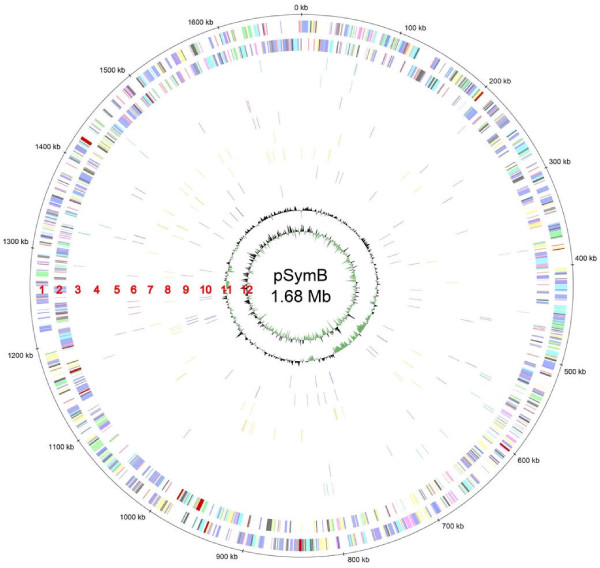
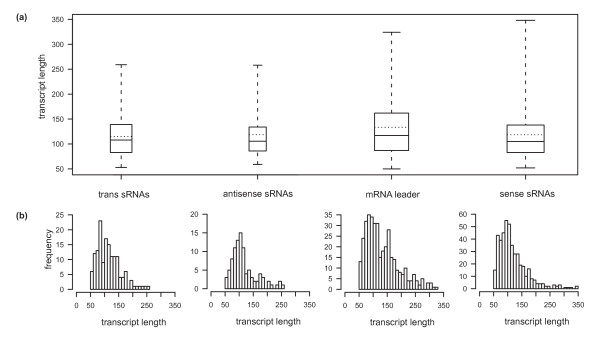
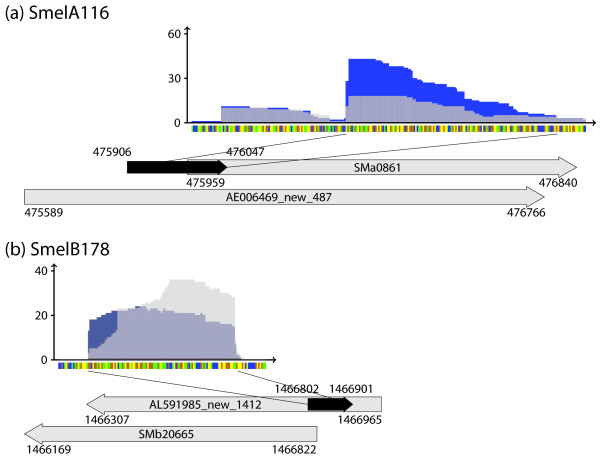
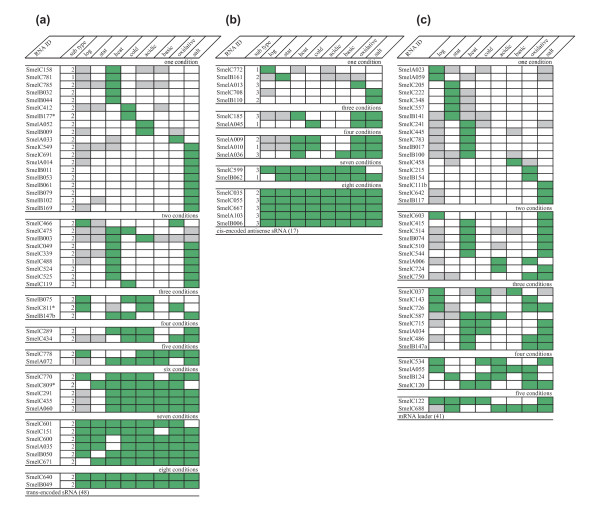
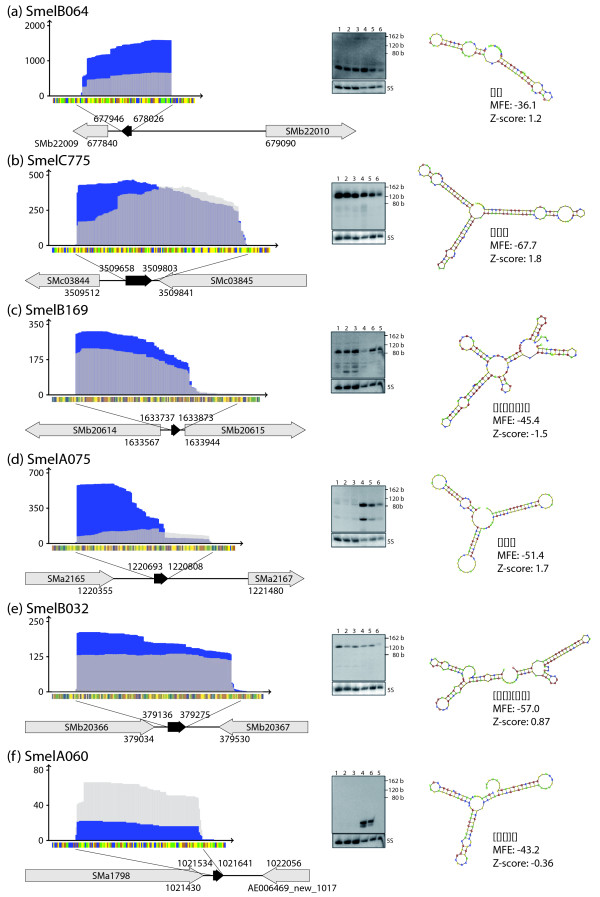
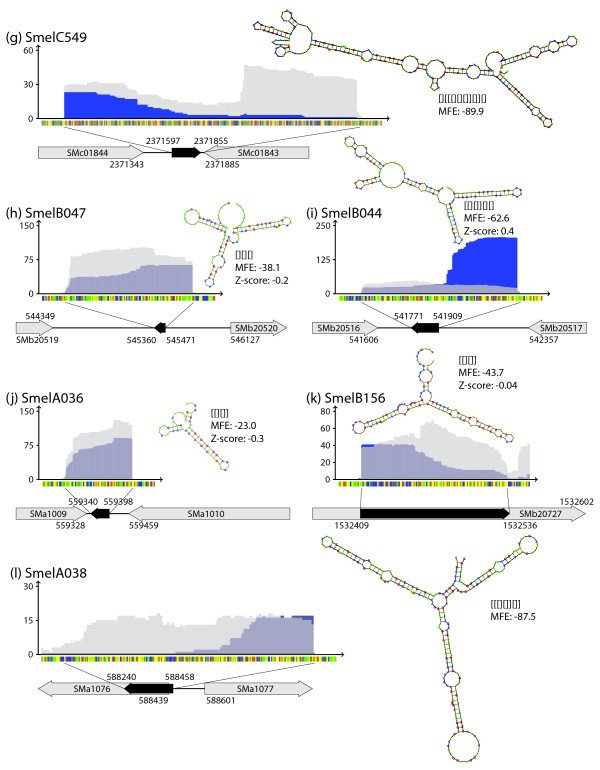
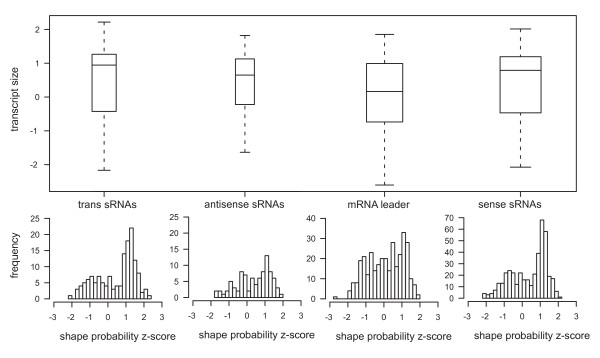
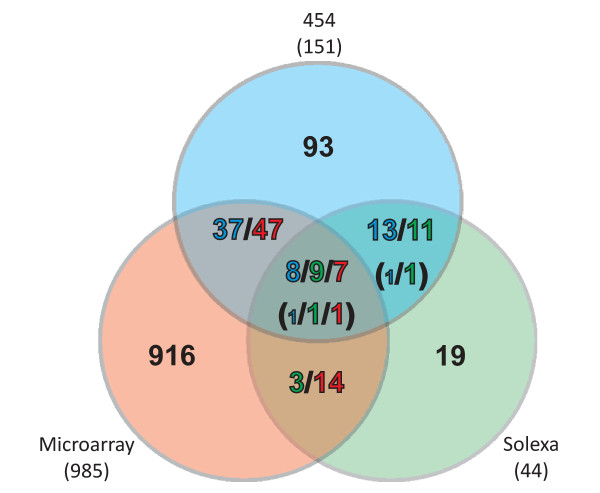
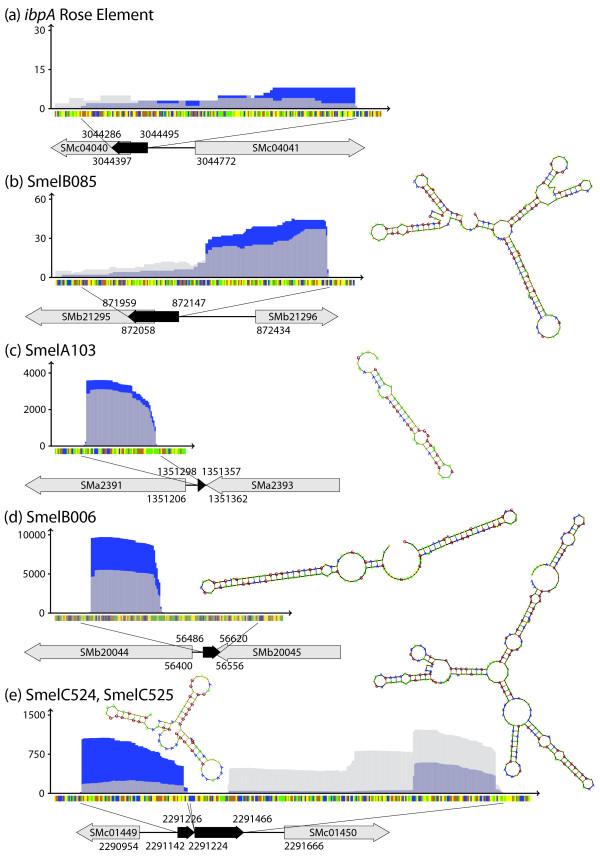
Results: A total of 1,125 sRNA candidates that were classified as trans-encoded sRNAs (173), cis-encoded antisense sRNAs (117), mRNA leader transcripts (379), and sense sRNAs overlapping coding regions (456) were identified in a size range of 50 to 348 nucleotides. Among these were transcripts corresponding to 82 previously reported sRNA candidates. Enrichment for RNAs with primary 5'-ends prior to sequencing of cDNAs suggested transcriptional start sites corresponding to 466 predicted sRNA regions. The consensus sigma70 promoter motif CTTGAC-N17-CTATAT was found upstream of 101 sRNA candidates. Expression patterns derived from microarray hybridizations provided further information on conditions of expression of a number of sRNA candidates. Furthermore, GenBank, EMBL, DDBJ, PDB, and Rfam databases were searched for homologs of the sRNA candidates identified in this study. Searching Rfam family models with over 1,000 sRNA candidates, re-discovered only those sequences from S. meliloti already known and stored in Rfam, whereas BLAST searches suggested a number of homologs in related alpha-proteobacteria.
Conclusions: The screening data suggests that in S. meliloti about 3% of the genes encode trans-encoded sRNAs and about 2% antisense transcripts. Thus, this first comprehensive screen for sRNAs applying deep sequencing in an alpha-proteobacterium shows that sRNAs also occur in high number in this group of bacteria.
Figures
Similar articles
-
Prediction of Sinorhizobium meliloti sRNA genes and experimental detection in strain 2011.BMC Genomics. 2008 Sep 16;9:416. doi: 10.1186/1471-2164-9-416. BMC Genomics. 2008. PMID: 18793445 Free PMC article.
-
Genome-wide profiling of Hfq-binding RNAs uncovers extensive post-transcriptional rewiring of major stress response and symbiotic regulons in Sinorhizobium meliloti.RNA Biol. 2014;11(5):563-79. doi: 10.4161/rna.28239. Epub 2014 Feb 26. RNA Biol. 2014. PMID: 24786641 Free PMC article.
-
Identification of differentially expressed small non-coding RNAs in the legume endosymbiont Sinorhizobium meliloti by comparative genomics.Mol Microbiol. 2007 Dec;66(5):1080-91. doi: 10.1111/j.1365-2958.2007.05978.x. Epub 2007 Oct 25. Mol Microbiol. 2007. PMID: 17971083 Free PMC article.
-
Insights into the noncoding RNome of nitrogen-fixing endosymbiotic α-proteobacteria.Mol Plant Microbe Interact. 2013 Feb;26(2):160-7. doi: 10.1094/MPMI-07-12-0186-CR. Mol Plant Microbe Interact. 2013. PMID: 22991999 Review.
-
Riboregulation in bacteria: From general principles to novel mechanisms of the trp attenuator and its sRNA and peptide products.Wiley Interdiscip Rev RNA. 2022 May;13(3):e1696. doi: 10.1002/wrna.1696. Epub 2021 Oct 14. Wiley Interdiscip Rev RNA. 2022. PMID: 34651439 Review.
Cited by
-
Quantitative proteomic analysis of the Hfq-regulon in Sinorhizobium meliloti 2011.PLoS One. 2012;7(10):e48494. doi: 10.1371/journal.pone.0048494. Epub 2012 Oct 30. PLoS One. 2012. PMID: 23119037 Free PMC article.
-
Transcription attenuation-derived small RNA rnTrpL regulates tryptophan biosynthesis gene expression in trans.Nucleic Acids Res. 2019 Jul 9;47(12):6396-6410. doi: 10.1093/nar/gkz274. Nucleic Acids Res. 2019. PMID: 30993322 Free PMC article.
-
Genome-wide transcriptome analysis of the plant pathogen Xanthomonas identifies sRNAs with putative virulence functions.Nucleic Acids Res. 2012 Mar;40(5):2020-31. doi: 10.1093/nar/gkr904. Epub 2011 Nov 12. Nucleic Acids Res. 2012. PMID: 22080557 Free PMC article.
-
Functional Genomics Approaches to Studying Symbioses between Legumes and Nitrogen-Fixing Rhizobia.High Throughput. 2018 May 18;7(2):15. doi: 10.3390/ht7020015. High Throughput. 2018. PMID: 29783718 Free PMC article. Review.
-
Synthetase of the methyl donor S-adenosylmethionine from nitrogen-fixing α-rhizobia can bind functionally diverse RNA species.RNA Biol. 2021 Aug;18(8):1111-1123. doi: 10.1080/15476286.2020.1829365. Epub 2020 Oct 10. RNA Biol. 2021. PMID: 33043803 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
