

Mitochondrial quality control and neurological disease: an emerging connection
- PMID: 20398440
- PMCID: PMC2871738
- DOI: 10.1017/S1462399410001456
Mitochondrial quality control and neurological disease: an emerging connection
Abstract
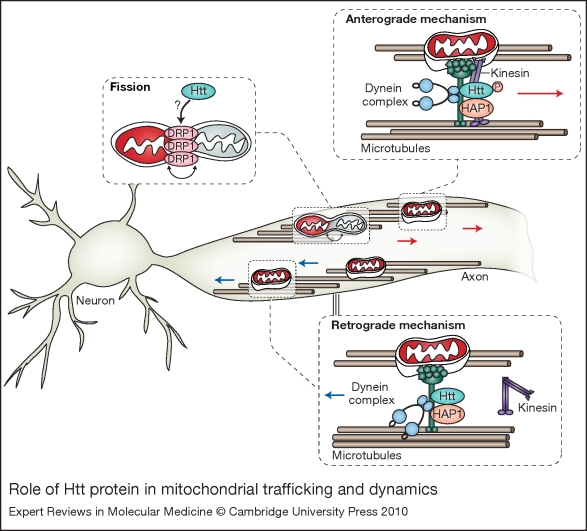
The human brain is a highly complex organ with remarkable energy demands. Although it represents only 2% of the total body weight, it accounts for 20% of all oxygen consumption, reflecting its high rate of metabolic activity. Mitochondria have a crucial role in the supply of energy to the brain. Consequently, their deterioration can have important detrimental consequences on the function and plasticity of neurons, and is thought to have a pivotal role in ageing and in the pathogenesis of several neurological disorders. Owing to their inherent physiological functions, mitochondria are subjected to particularly high levels of stress and have evolved specific molecular quality-control mechanisms to maintain the mitochondrial components. Here, we review some of the most recent advances in the understanding of mitochondrial stress-control pathways, with a particular focus on how defects in such pathways might contribute to neurodegenerative disease.
Figures




References
-
- Lane N. Power, Sex, Suicide: mitochondria and the meaning of life. Oxford University Press; Oxford: 2005.
-
- Chan D.C.. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–1252. - PubMed
-
- Taylor S.W., Fahy E.. Ghosh S.S.. Global organellar proteomics. Trends in Biotechnology. 2003;21:82–88. and . - PubMed
-
- Wang Y.. Bogenhagen D.F.. Human mitochondrial DNA nucleoids are linked to protein folding machinery and metabolic enzymes at the mitochondrial inner membrane. Journal of Biological Chemistry. 2006;281:25791–25802. and . - PubMed
Further reading, resources and contacts
Books
-
- Lane N. Power, Sex, Suicide: Mitochondria and the Meaning of Life. OUP; Oxford: 2005.
-
A provocative book full of interesting concepts, including links between ageing and neurodegenerative disease.
-
- Gibson G.E., Ratan R.R.. Flint Beal M.. Mitochondria and Oxidative Stress in Neurodegenerative Disorders. Annals of the New York Academy of Sciences 2008 and . eds ( - PubMed
-
A combination of basic and clinical research to give the reader the most current information on aspects of mitochondrial function linked to age-related neurodegenerative diseases and their treatment.
Websites
-
- http://www.alz.org http://www.alz.org
-
A database of Drosophila melanogaster nuclear genes encoding mitochondrial proteins can be found at:
-
- http://www2.ba.itb.cnr.it/MitoDrome http://www2.ba.itb.cnr.it/MitoDrome
-
The Whitehead Institute Video Gallery has lectures from researchers at the Whitehead Institute. It includes several interesting lectures by Susan Lindquist on protein misfolding and neurodegenerative diseases as well as David Sabatini on growth control pathways:
-
- www.wi.mit.edu/news/video_gallery www.wi.mit.edu/news/video_gallery
-
The Encyclopedia of Neuroscience explores a wide variety of topics related to different areas of neuroscience. It is a very useful tool, well written and easily accessible:
-
- www.sciencedirect.com/science/referenceworks/9780080450469 www.sciencedirect.com/science/referenceworks/9780080450469
-
The Allen Mouse Brain Atlas is an interactive, genome-wide image database of gene expression. It also includes information regarding the cortex of the human brain:
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
