

Dynameomics: a comprehensive database of protein dynamics
- PMID: 20399180
- PMCID: PMC2892689
- DOI: 10.1016/j.str.2010.01.012
Dynameomics: a comprehensive database of protein dynamics
Abstract
The dynamic behavior of proteins is important for an understanding of their function and folding. We have performed molecular dynamics simulations of the native state and unfolding pathways of over 2000 protein/peptide systems (approximately 11,000 independent simulations) representing the majority of folds in globular proteins. These data are stored and organized using an innovative database approach, which can be mined to obtain both general and specific information about the dynamics and folding/unfolding of proteins, relevant subsets thereof, and individual proteins. Here we describe the project in general terms and the type of information contained in the database. Then we provide examples of mining the database for information relevant to protein folding, structure building, the effect of single-nucleotide polymorphisms, and drug design. The native state simulation data and corresponding analyses for the 100 most populated metafolds, together with related resources, are publicly accessible through http://www.dynameomics.org.
Copyright 2010 Elsevier Ltd. All rights reserved.
Figures
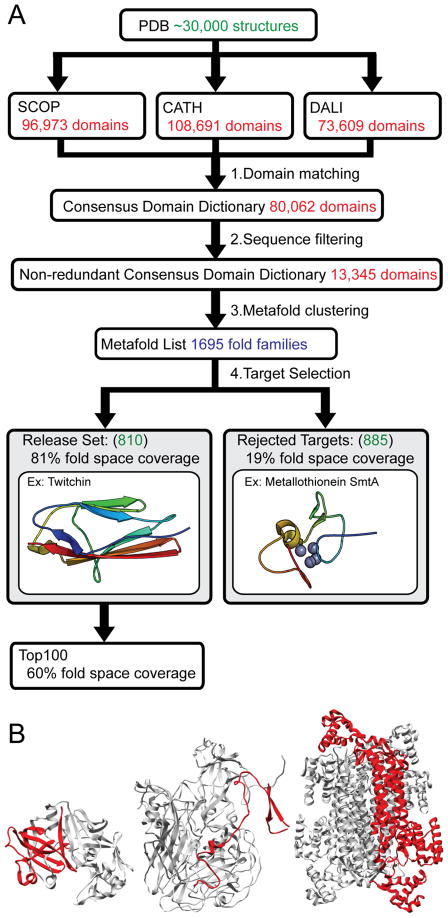
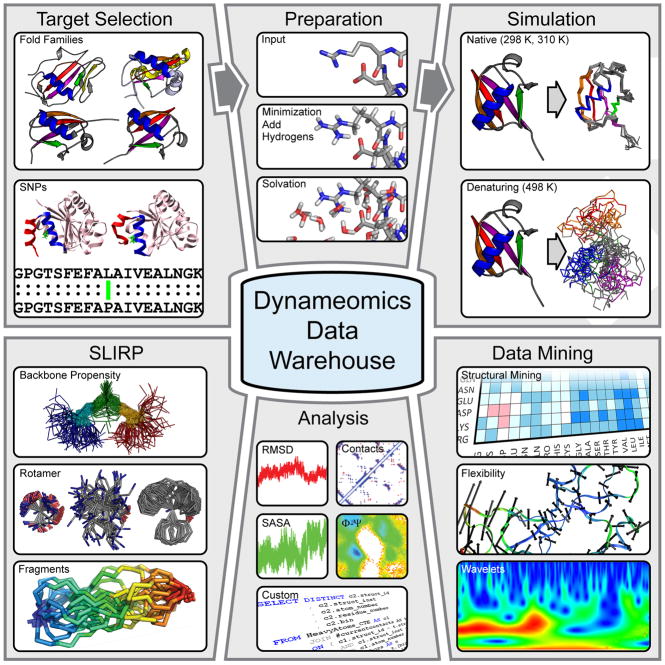
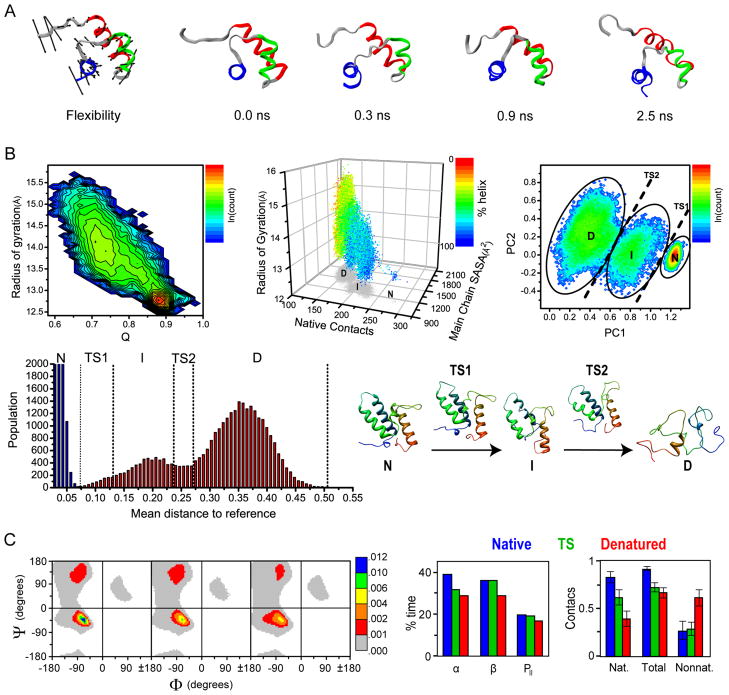
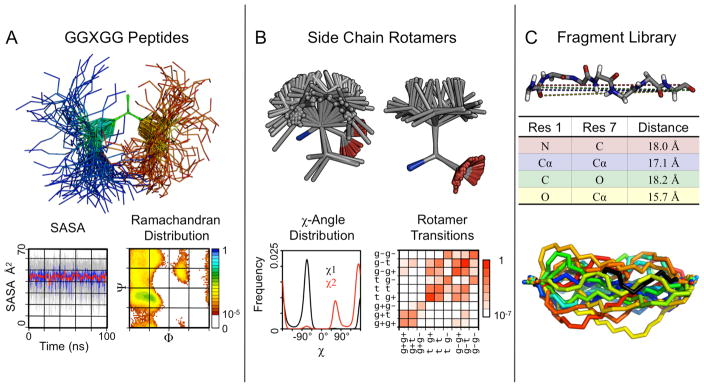
Comment in
-
Making membrane proteins for structures: a trillion tiny tweaks.Nat Methods. 2010 Jun;7(6):429-34. doi: 10.1038/nmeth0610-429. Nat Methods. 2010. PMID: 20508636 No abstract available.
-
A database of dynamics.Nat Methods. 2010 Jun;7(6):426. doi: 10.1038/nmeth0610-426. Nat Methods. 2010. PMID: 20524218
Similar articles
-
Dynameomics: a consensus view of the protein unfolding/folding transition state ensemble across a diverse set of protein folds.Biophys J. 2009 Dec 2;97(11):2958-66. doi: 10.1016/j.bpj.2009.09.012. Biophys J. 2009. PMID: 19948125 Free PMC article.
-
Dynameomics: mass annotation of protein dynamics and unfolding in water by high-throughput atomistic molecular dynamics simulations.Protein Eng Des Sel. 2008 Jun;21(6):353-68. doi: 10.1093/protein/gzn011. Epub 2008 Apr 14. Protein Eng Des Sel. 2008. PMID: 18411224
-
Dynameomics: design of a computational lab workflow and scientific data repository for protein simulations.Protein Eng Des Sel. 2008 Jun;21(6):369-77. doi: 10.1093/protein/gzn012. Epub 2008 Apr 14. Protein Eng Des Sel. 2008. PMID: 18411223
-
Protein folds and protein folding.Protein Eng Des Sel. 2011 Jan;24(1-2):11-9. doi: 10.1093/protein/gzq096. Epub 2010 Nov 3. Protein Eng Des Sel. 2011. PMID: 21051320 Free PMC article. Review.
-
Proteins in vacuo: denaturing and folding mechanisms studied with computer-simulated molecular dynamics.Mass Spectrom Rev. 2001 Nov-Dec;20(6):402-22. doi: 10.1002/mas.10012. Mass Spectrom Rev. 2001. PMID: 11997946 Review.
Cited by
-
Tumorigenic p53 mutants undergo common structural disruptions including conversion to α-sheet structure.Protein Sci. 2020 Sep;29(9):1983-1999. doi: 10.1002/pro.3921. Epub 2020 Aug 17. Protein Sci. 2020. PMID: 32715544 Free PMC article.
-
Shared unfolding pathways of unrelated immunoglobulin-like β-sandwich proteins.Protein Eng Des Sel. 2019 Dec 31;32(7):331-345. doi: 10.1093/protein/gzz040. Protein Eng Des Sel. 2019. PMID: 31868211 Free PMC article.
-
Remote thioredoxin recognition using evolutionary conservation and structural dynamics.Structure. 2011 Apr 13;19(4):461-70. doi: 10.1016/j.str.2011.02.007. Structure. 2011. PMID: 21481770 Free PMC article.
-
The role of α-sheet structure in amyloidogenesis: characterization and implications.Open Biol. 2022 Nov;12(11):220261. doi: 10.1098/rsob.220261. Epub 2022 Nov 23. Open Biol. 2022. PMID: 36416010 Free PMC article. Review.
-
Designed α-sheet peptides inhibit amyloid formation by targeting toxic oligomers.Elife. 2014 Jul 15;3:e01681. doi: 10.7554/eLife.01681. Elife. 2014. PMID: 25027691 Free PMC article.
References
-
- Beck DAC, Alonso DOV, Daggett V. In lucem molecular mechanics. Seattle, WA: University of Washington; 2000–2010.
-
- Beck DAC, Armen RS, Daggett V. Cutoff size need not strongly influence molecular dynamics results for solvated polypeptides. Biochem. 2005;44:609–616. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
