

Structure of concatenated HAMP domains provides a mechanism for signal transduction
- PMID: 20399181
- PMCID: PMC2892831
- DOI: 10.1016/j.str.2010.01.013
Structure of concatenated HAMP domains provides a mechanism for signal transduction
Abstract
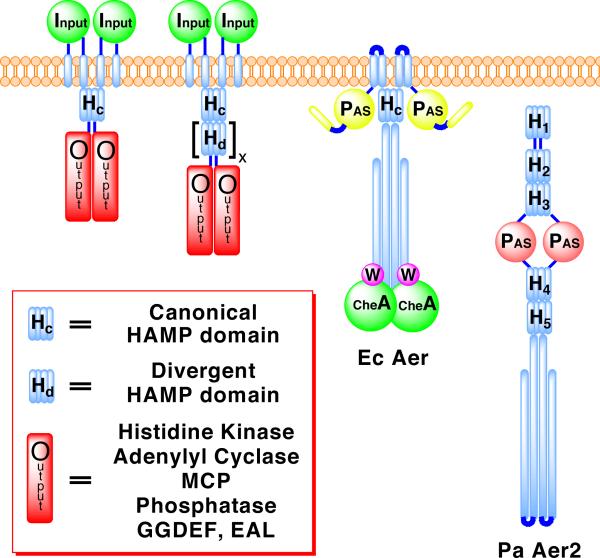
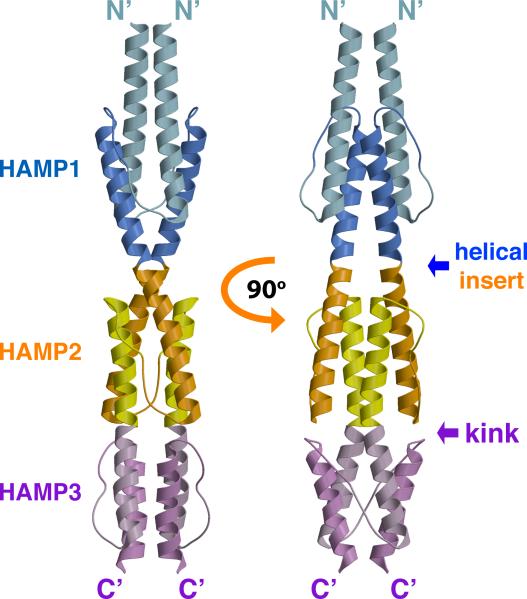
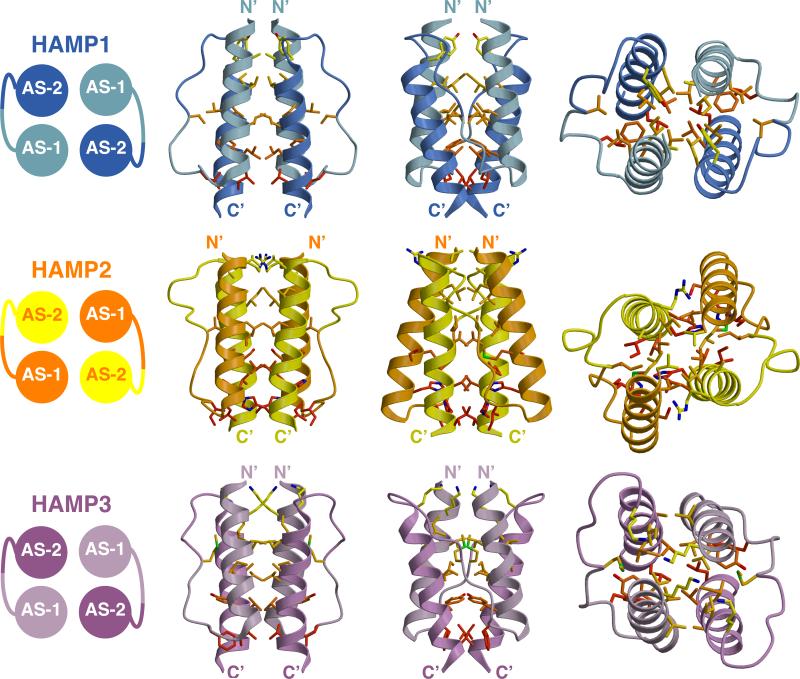
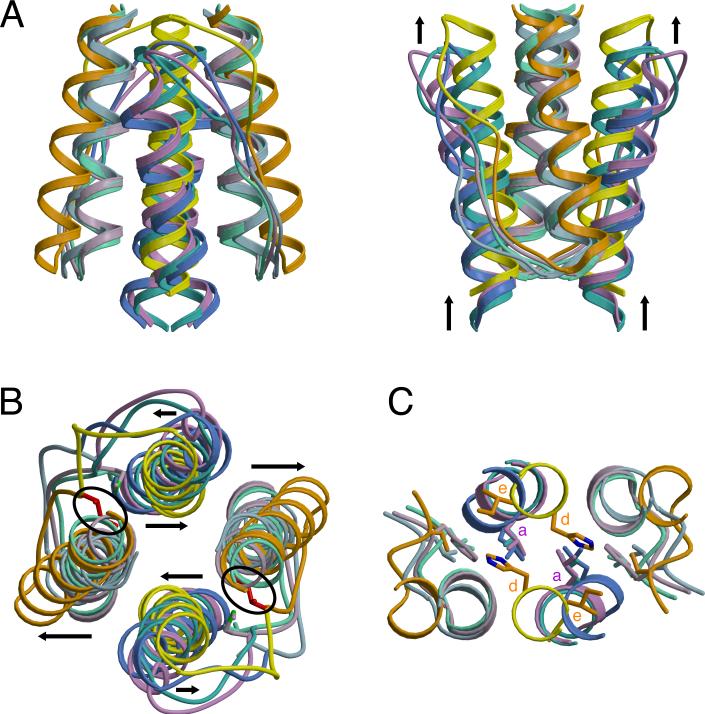
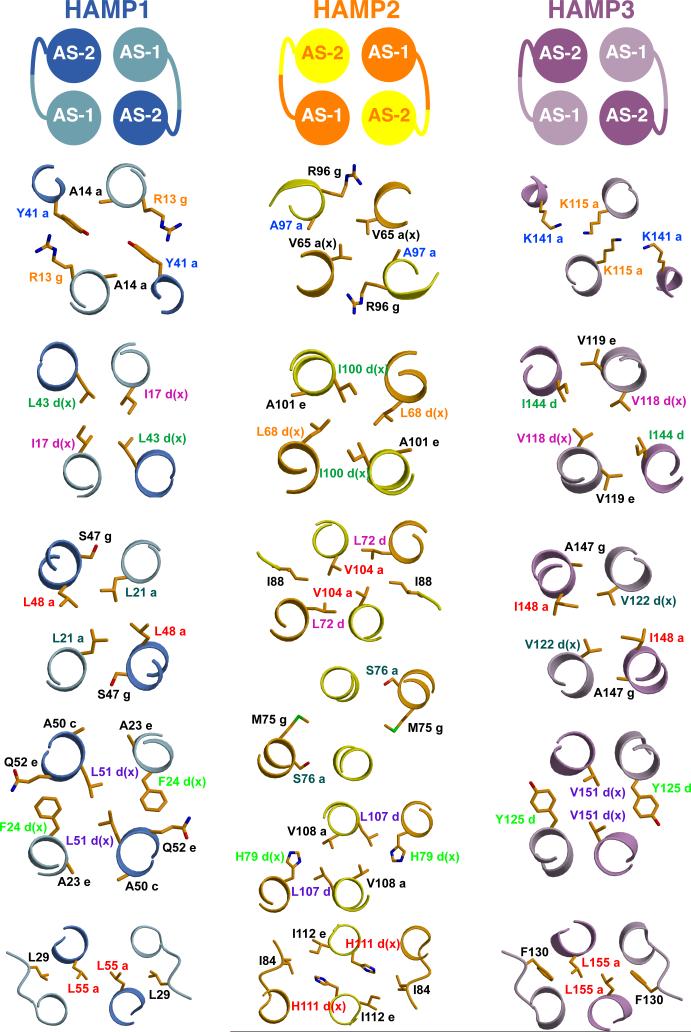
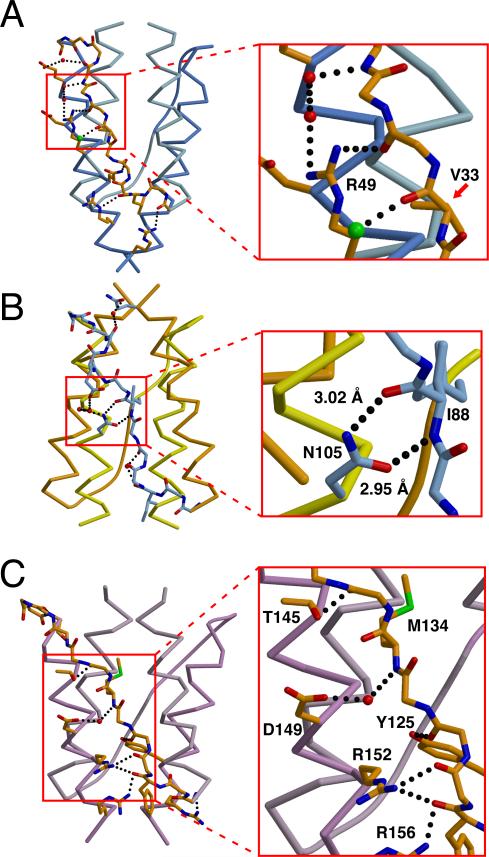
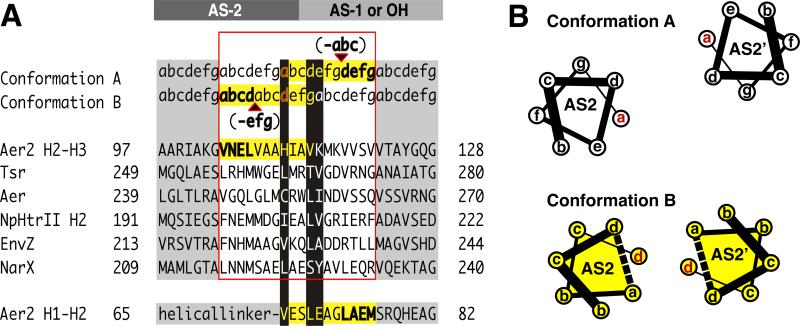
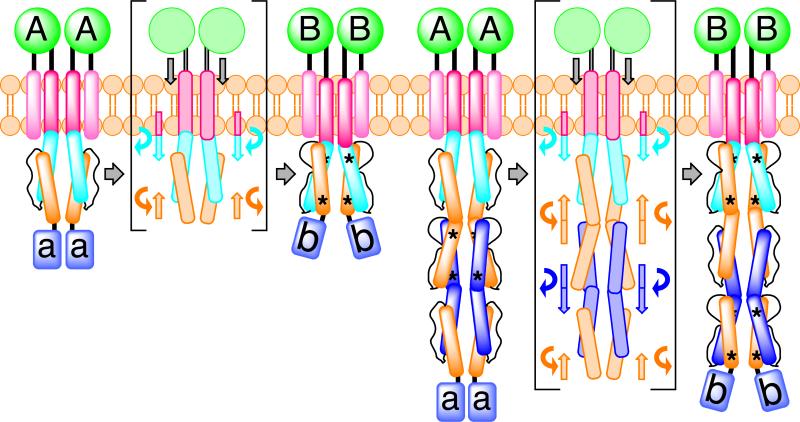
HAMP domains are widespread prokaryotic signaling modules found as single domains or poly-HAMP chains in both transmembrane and soluble proteins. The crystal structure of a three-unit poly-HAMP chain from the Pseudomonas aeruginosa soluble receptor Aer2 defines a universal parallel four-helix bundle architecture for diverse HAMP domains. Two contiguous domains integrate to form a concatenated di-HAMP structure. The three HAMP domains display two distinct conformations that differ by changes in helical register, crossing angle, and rotation. These conformations are stabilized by different subsets of conserved residues. Known signals delivered to HAMP would be expected to switch the relative stability of the two conformations and the position of a coiled-coil phase stutter at the junction with downstream helices. We propose that the two conformations represent opposing HAMP signaling states and suggest a signaling mechanism whereby HAMP domains interconvert between the two states, which alternate down a poly-HAMP chain.
Copyright 2010 Elsevier Ltd. All rights reserved.
Figures

References
-
- Ambroggio XI, Kuhlman B. Design of protein conformational switches. Current Opinion in Structural Biology. 2006;16:525–530. - PubMed
-
- Aravind L, Ponting CP. The cytoplasmic helical linker domain of receptor histidine kinase and methyl-accepting proteins is common to many prokaryotic signalling proteins. Fems Microbiology Letters. 1999;176:111–116. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
