

A CD8+ T cell transcription signature predicts prognosis in autoimmune disease
- PMID: 20400961
- PMCID: PMC3504359
- DOI: 10.1038/nm.2130
A CD8+ T cell transcription signature predicts prognosis in autoimmune disease
Abstract
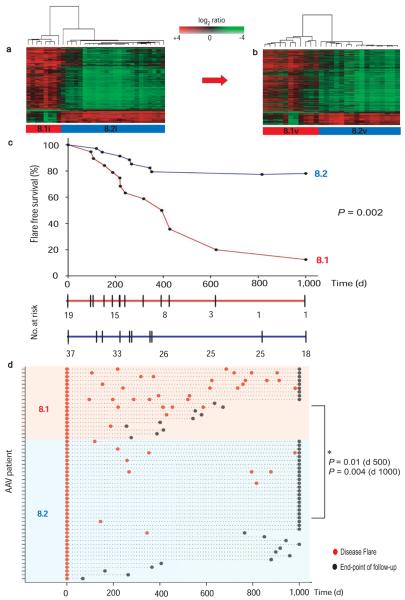
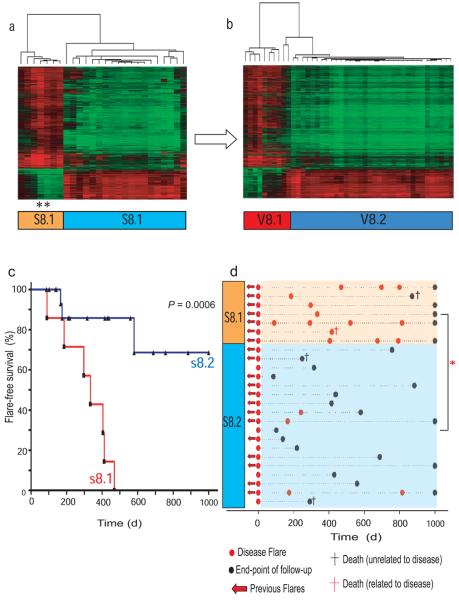
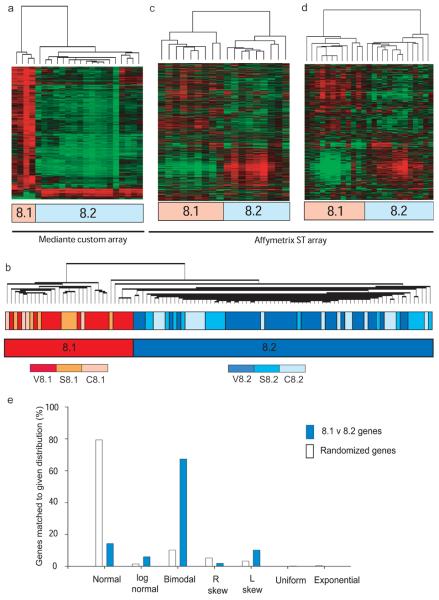
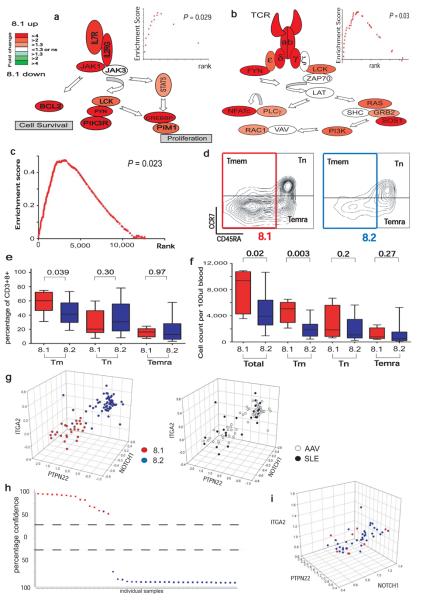
Autoimmune diseases are common and debilitating, but their severe manifestations could be reduced if biomarkers were available to allow individual tailoring of potentially toxic immunosuppressive therapy. Gene expression-based biomarkers facilitating such tailoring of chemotherapy in cancer, but not autoimmunity, have been identified and translated into clinical practice. We show that transcriptional profiling of purified CD8(+) T cells, which avoids the confounding influences of unseparated cells, identifies two distinct subject subgroups predicting long-term prognosis in two autoimmune diseases, antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV), a chronic, severe disease characterized by inflammation of medium-sized and small blood vessels, and systemic lupus erythematosus (SLE), characterized by autoantibodies, immune complex deposition and diverse clinical manifestations ranging from glomerulonephritis to neurological dysfunction. We show that the subset of genes defining the poor prognostic group is enriched for genes involved in the interleukin-7 receptor (IL-7R) pathway and T cell receptor (TCR) signaling and those expressed by memory T cells. Furthermore, the poor prognostic group is associated with an expanded CD8(+) T cell memory population. These subgroups, which are also found in the normal population and can be identified by measuring expression of only three genes, raise the prospect of individualized therapy and suggest new potential therapeutic targets in autoimmunity.
Figures
Comment in
-
Biomarkers: New prognostic biomarker for autoimmune diseases.Nat Rev Rheumatol. 2010 Jul;6(7):380. doi: 10.1038/nrrheum.2010.90. Nat Rev Rheumatol. 2010. PMID: 20614501 No abstract available.
References
-
- Golub TR, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–7. - PubMed
-
- van't Veer LJ, Bernards R. Enabling personalized cancer medicine through analysis of gene-expression patterns. Nature. 2008;452:564–70. - PubMed
-
- Lane SE, Watts RA, Shepstone L, Scott DG. Primary systemic vasculitis: clinical features and mortality. QJM. 2005;98:97–111. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
