

Testicular postgenomics: targeting the regulation of spermatogenesis
- PMID: 20403865
- PMCID: PMC2871924
- DOI: 10.1098/rstb.2009.0294
Testicular postgenomics: targeting the regulation of spermatogenesis
Abstract
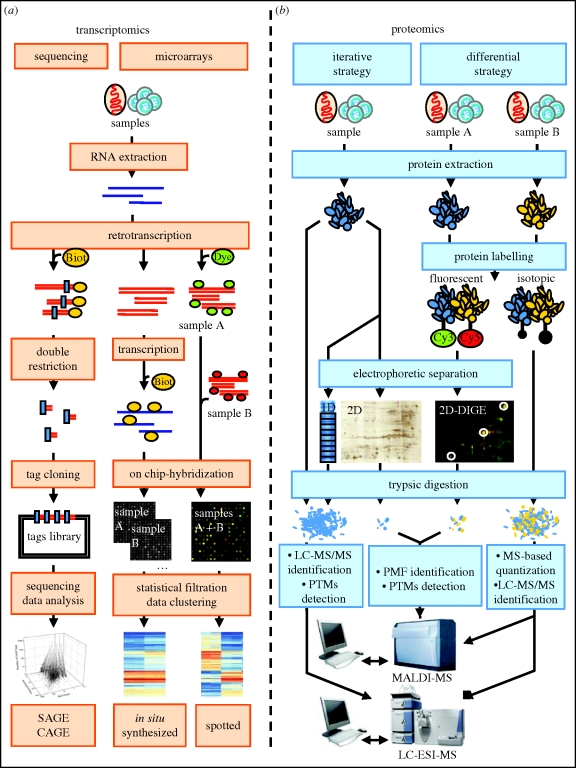
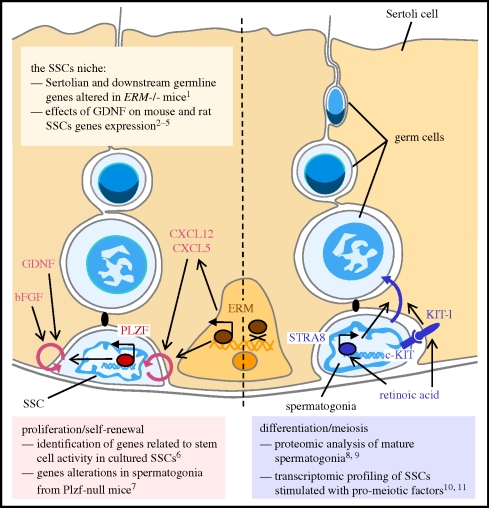
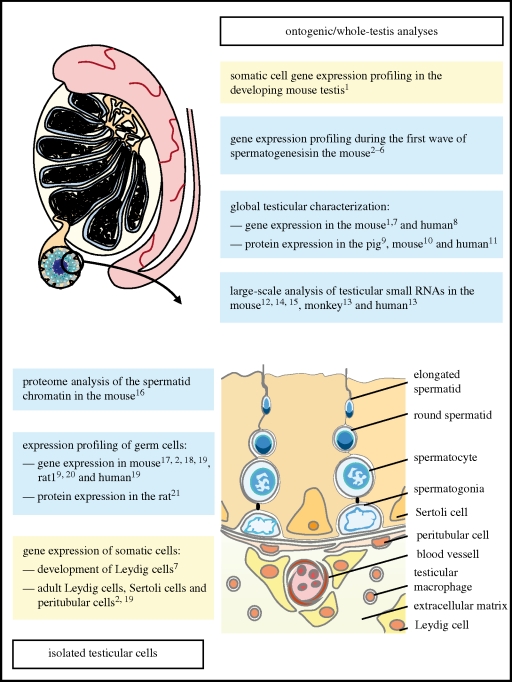
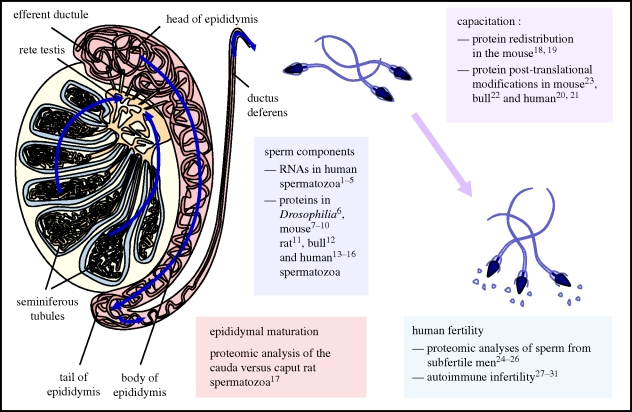
Sperm are, arguably, the most differentiated cells produced within the body of any given species. This is owing to the fact that spermatogenesis is an intricate and highly specialized process evolved to suit the individual particularities of each sexual species. Despite a vast diversity in method, the aim of spermatogenesis is always the same, the idealized transmission of genetic patrimony. Towards this goal certain requirements must always be met, such as a relative twofold reduction in ploidy, repackaging of the chromatin for transport and specialized enhancements for cell motility, recognition and fusion. In the past 20 years, the study of molecular networks coordinating male germ cell development, particularly in mammals, has become more and more facilitated thanks to large-scale analyses of genome expression. Such postgenomic endeavors have generated landscapes of data for both fundamental and clinical reproductive biology. Continuous, large-scale integration analyses of these datasets are undertaken which provide access to very precise information on a myriad of biomolecules. This review presents commonly used transcriptomic and proteomic workflows applied to various testicular germ cell studies. We will also provide a general overview of the technical possibilities available to reproductive genomic biologists, noting the advantages and drawbacks of each technique.
Figures

References
-
- Aebersold R., Mann M.2003Mass spectrometry-based proteomics. Nature 422, 198–207 (doi:10.1038/nature01511) - DOI - PubMed
-
- Almstrup K., Nielsen J. E., Hansen M. A., Tanaka M., Skakkebaek N. E., Leffers H.2004Analysis of cell-type-specific gene expression during mouse spermatogenesis. Biol. Reprod. 70, 1751–1761 (doi:10.1095/biolreprod.103.026575) - DOI - PubMed
-
- Anderson N. L., Anderson N. G.1998Proteome and proteomics: new technologies, new concepts, and new words. Electrophoresis 19, 1853–1861 (doi:10.1002/elps.1150191103) - DOI - PubMed
-
- Andrabi S. M.2007Mammalian sperm chromatin structure and assessment of DNA fragmentation. J. Assist. Reprod. Genet. 24, 561–569 (doi:10.1007/s10815-007-9177-y) - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
