

Chromosome missegregation causes colon cancer by APC loss of heterozygosity
- PMID: 20404532
- PMCID: PMC2963855
- DOI: 10.4161/cc.9.9.11314
Chromosome missegregation causes colon cancer by APC loss of heterozygosity
Abstract
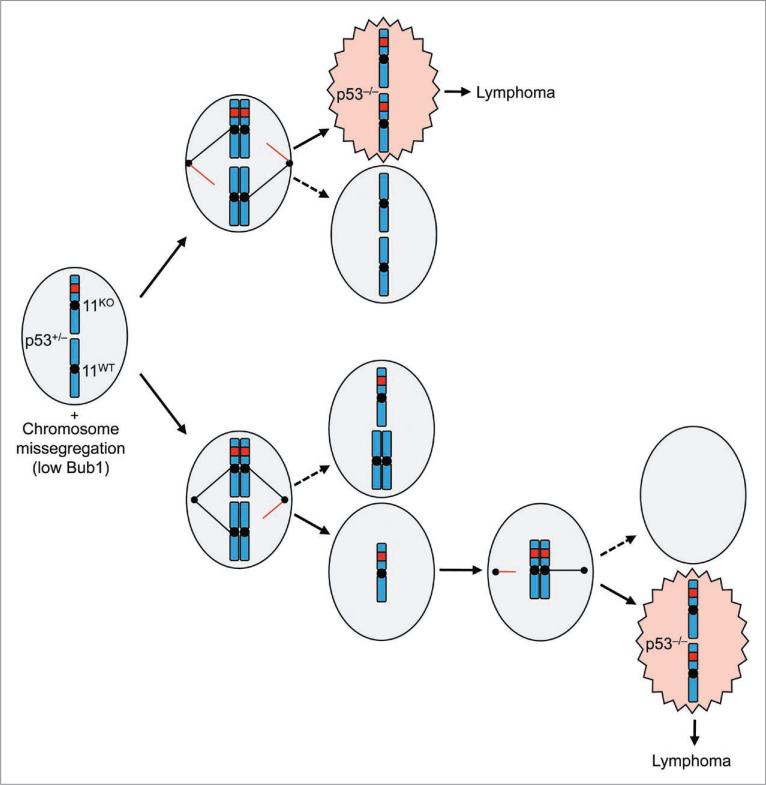
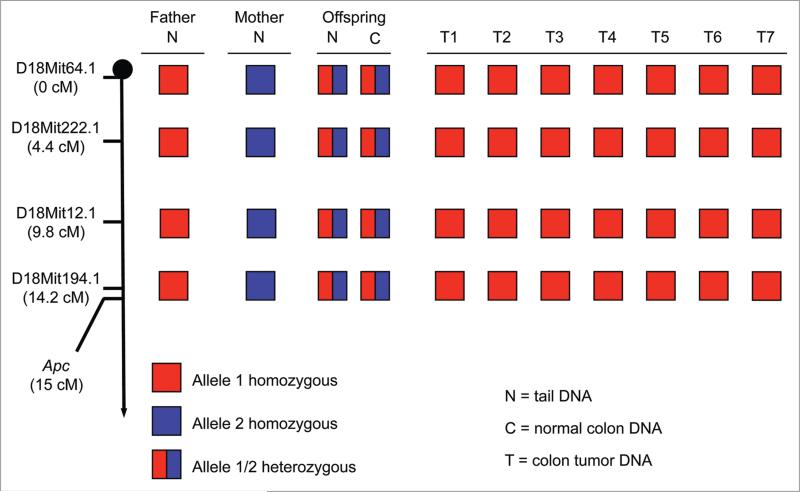
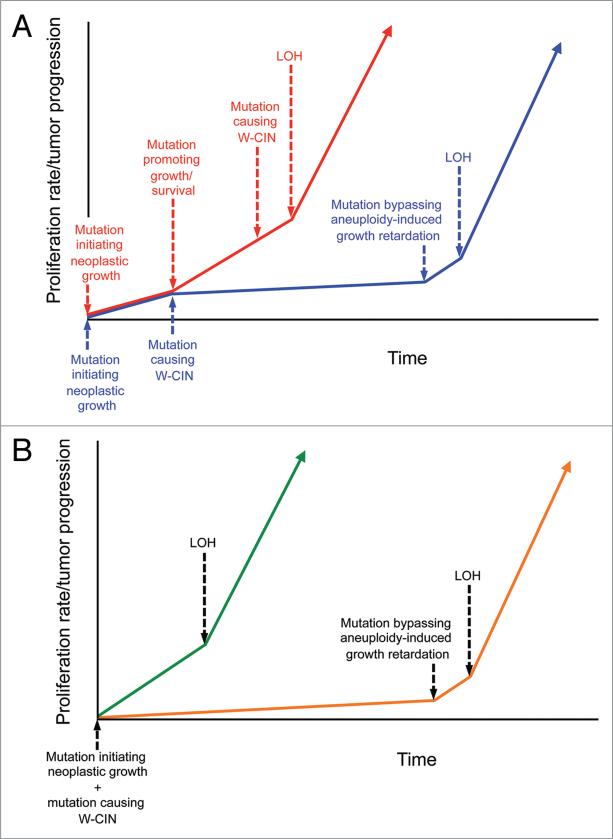
A longstanding hypothesis in the field of cancer biology is that aneuploidy causes cancer by promoting loss of chromosomes that contain tumor suppressor genes. By crossing aneuploidyprone Bub1 hypomorphic mice onto a heterozygous null background for p53, we provided conclusive evidence for this idea.(1) Surprisingly, the tumors that developed in this model had not just lost the chromosome 11 copy harboring wild-type p53, but had also gained an extra copy of chromosome 11 bearing the p53 null allele. Here we report that a similar chromosome-reshuffling blueprint drives colonic tumorigenesis in Bub1 hypomorphic mice that are heterozygous for Apc(Min), but now involving chromosome 18. These extended studies highlight that in order for whole chromosome instability to drive tumorigenesis, it needs to establish tumor suppressor gene loss of heterozygosity while retaining two copies of the other genes on the chromosome. Additional restrictions seem to apply to whole chromosome instability as a cancer causing mechanism, which will be discussed in this paper.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous