

Cellular and molecular pathways to myocardial necrosis and replacement fibrosis
- PMID: 20405318
- PMCID: PMC2920342
- DOI: 10.1007/s10741-010-9169-3
Cellular and molecular pathways to myocardial necrosis and replacement fibrosis
Abstract
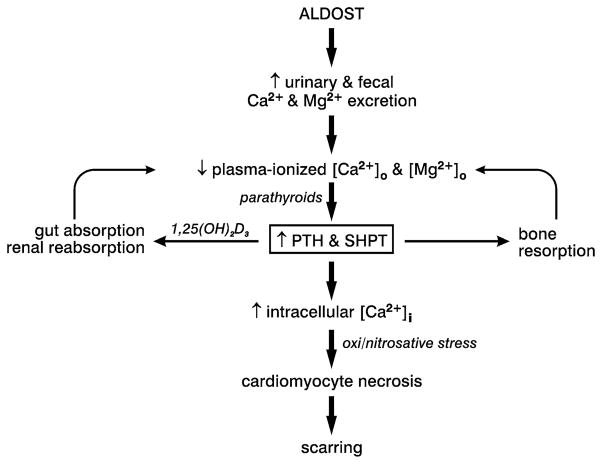
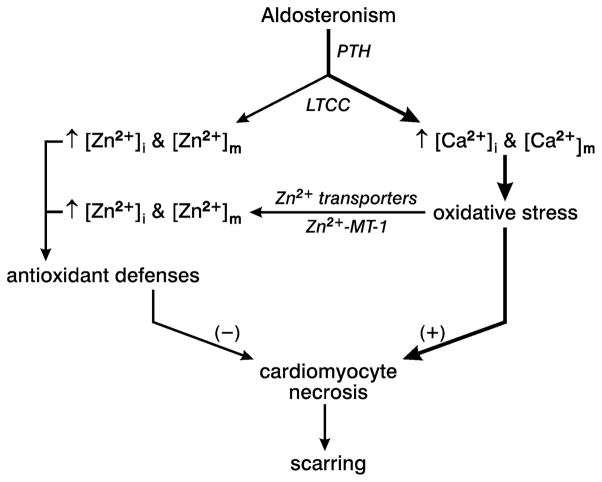
Fibrosis is a fundamental component of the adverse structural remodeling of myocardium present in the failing heart. Replacement fibrosis appears at sites of previous cardiomyocyte necrosis to preserve the structural integrity of the myocardium, but not without adverse functional consequences. The extensive nature of this microscopic scarring suggests cardiomyocyte necrosis is widespread and the loss of these contractile elements, combined with fibrous tissue deposition in the form of a stiff in-series and in-parallel elastic elements, contributes to the progressive failure of this normally efficient muscular pump. Cellular and molecular studies into the signal-transducer-effector pathway involved in cardiomyocyte necrosis have identified the crucial pathogenic role of intracellular Ca2+ overloading and subsequent induction of oxidative stress, predominantly confined within its mitochondria, to be followed by the opening of the mitochondrial permeability transition pore that leads to the destruction of these organelles and cells. It is now further recognized that Ca2+ overloading of cardiac myocytes and mitochondria serves as a prooxidant and which is counterbalanced by an intrinsically coupled Zn2+ entry serving as antioxidant. The prospect of raising antioxidant defenses by increasing intracellular Zn2+ with adjuvant nutriceuticals can, therefore, be preferentially exploited to uncouple this intrinsically coupled Ca2+ - Zn2+ dyshomeostasis. Hence, novel yet simple cardioprotective strategies may be at hand that deserve to be further explored.
Figures
Similar articles
-
Fibrosis in hypertensive heart disease: molecular pathways and cardioprotective strategies.J Hypertens. 2010 Sep;28 Suppl 1(Suppl 1):S25-32. doi: 10.1097/01.hjh.0000388491.35836.d2. J Hypertens. 2010. PMID: 20823713 Free PMC article. Review.
-
Congestive heart failure: where homeostasis begets dyshomeostasis.J Cardiovasc Pharmacol. 2010 Sep;56(3):320-8. doi: 10.1097/FJC.0b013e3181ed064f. J Cardiovasc Pharmacol. 2010. PMID: 20588190 Free PMC article.
-
From aldosteronism to oxidative stress: the role of excessive intracellular calcium accumulation.Hypertens Res. 2010 Nov;33(11):1091-101. doi: 10.1038/hr.2010.159. Epub 2010 Sep 9. Hypertens Res. 2010. PMID: 20827279 Review.
-
Myocardial remodeling in low-renin hypertension: molecular pathways to cellular injury in relative aldosteronism.Curr Hypertens Rep. 2009 Dec;11(6):412-20. doi: 10.1007/s11906-009-0071-0. Curr Hypertens Rep. 2009. PMID: 19895752 Free PMC article. Review.
-
Mitochondria play a central role in nonischemic cardiomyocyte necrosis: common to acute and chronic stressor states.Pflugers Arch. 2012 Jul;464(1):123-31. doi: 10.1007/s00424-012-1079-x. Epub 2012 Feb 11. Pflugers Arch. 2012. PMID: 22328074 Free PMC article. Review.
Cited by
-
Metformin Attenuates Postinfarction Myocardial Fibrosis and Inflammation in Mice.Int J Mol Sci. 2021 Aug 30;22(17):9393. doi: 10.3390/ijms22179393. Int J Mol Sci. 2021. PMID: 34502314 Free PMC article.
-
Mst1 inhibition rescues β1-adrenergic cardiomyopathy by reducing myocyte necrosis and non-myocyte apoptosis rather than myocyte apoptosis.Basic Res Cardiol. 2015 Mar;110(2):7. doi: 10.1007/s00395-015-0461-1. Epub 2015 Jan 20. Basic Res Cardiol. 2015. PMID: 25600225 Free PMC article.
-
Proteome analysis of subsarcolemmal cardiomyocyte mitochondria: a comparison of different analytical platforms.Int J Mol Sci. 2014 May 26;15(6):9285-301. doi: 10.3390/ijms15069285. Int J Mol Sci. 2014. PMID: 24865490 Free PMC article.
-
Cardiospecific deletion of αE-catenin leads to heart failure and lethality in mice.Pflugers Arch. 2018 Oct;470(10):1485-1499. doi: 10.1007/s00424-018-2168-2. Epub 2018 Jun 20. Pflugers Arch. 2018. PMID: 29923116
-
Adult murine cardiomyocytes exhibit regenerative activity with cell cycle reentry through STAT3 in the healing process of myocarditis.Sci Rep. 2017 May 3;7(1):1407. doi: 10.1038/s41598-017-01426-8. Sci Rep. 2017. PMID: 28469272 Free PMC article.
References
-
- Beltrami CA, Finato N, Rocco M, Feruglio GA, Puricelli C, Cigola E, Quaini F, Sonnenblick EH, Olivetti G, Anversa P. Structural basis of end-stage failure in ischemic cardiomyopathy in humans. Circulation. 1994;89:151–163. - PubMed
-
- Cotran RS, Kumar V, Robbins SL. The heart. In: Cotran RS, Kumar V, Robbins SL, editors. Robbins pathologic basis of disease. 4. WB Saunders; Philadelphia: 1989. pp. 597–656.
-
- Li H, Ambade A, Re F. Cutting edge: necrosis activates the NLRP3 inflammasome. J Immunol. 2009;183:1528–1532. - PubMed
-
- Cohen I, Rider P, Carmi Y, Braiman A, Dotan S, White MR, Voronov E, Martin MU, Dinarello CA, Apte RN. Differential release of chromatin-bound IL-1α discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc Natl Acad Sci U S A. 2010;107:2574–2579. - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
