

The impact of genetic architecture on genome-wide evaluation methods
- PMID: 20407128
- PMCID: PMC2907189
- DOI: 10.1534/genetics.110.116855
The impact of genetic architecture on genome-wide evaluation methods
Abstract
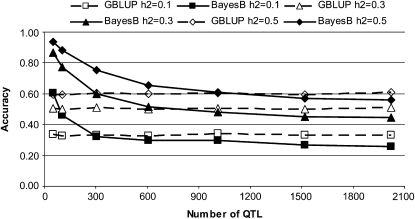
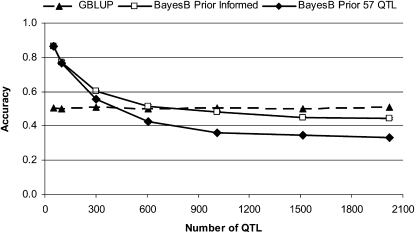
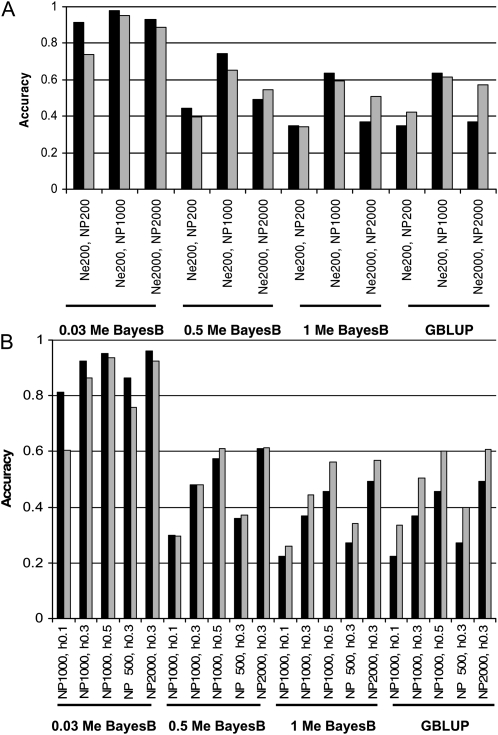
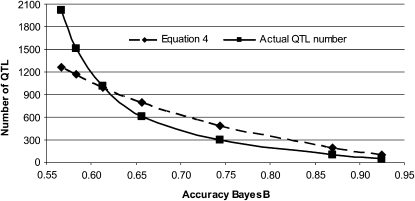
The rapid increase in high-throughput single-nucleotide polymorphism data has led to a great interest in applying genome-wide evaluation methods to identify an individual's genetic merit. Genome-wide evaluation combines statistical methods with genomic data to predict genetic values for complex traits. Considerable uncertainty currently exists in determining which genome-wide evaluation method is the most appropriate. We hypothesize that genome-wide methods deal differently with the genetic architecture of quantitative traits and genomes. A genomic linear method (GBLUP), and a genomic nonlinear Bayesian variable selection method (BayesB) are compared using stochastic simulation across three effective population sizes and a wide range of numbers of quantitative trait loci (N(QTL)). GBLUP had a constant accuracy, for a given heritability and sample size, regardless of N(QTL). BayesB had a higher accuracy than GBLUP when N(QTL) was low, but this advantage diminished as N(QTL) increased and when N(QTL) became large, GBLUP slightly outperformed BayesB. In addition, deterministic equations are extended to predict the accuracy of both methods and to estimate the number of independent chromosome segments (M(e)) and N(QTL). The predictions of accuracy and estimates of M(e) and N(QTL) were generally in good agreement with results from simulated data. We conclude that the relative accuracy of GBLUP and BayesB for a given number of records and heritability are highly dependent on M(e,) which is a property of the target genome, as well as the architecture of the trait (N(QTL)).
Figures
Similar articles
-
Across population genomic prediction scenarios in which Bayesian variable selection outperforms GBLUP.BMC Genet. 2015 Dec 23;16:146. doi: 10.1186/s12863-015-0305-x. BMC Genet. 2015. PMID: 26698836 Free PMC article.
-
Mixture models detect large effect QTL better than GBLUP and result in more accurate and persistent predictions.J Anim Sci Biotechnol. 2016 Feb 11;7:7. doi: 10.1186/s40104-016-0066-z. eCollection 2016. J Anim Sci Biotechnol. 2016. PMID: 26870325 Free PMC article.
-
Empirical and deterministic accuracies of across-population genomic prediction.Genet Sel Evol. 2015 Feb 6;47(1):5. doi: 10.1186/s12711-014-0086-0. Genet Sel Evol. 2015. PMID: 25885467 Free PMC article.
-
Different models of genetic variation and their effect on genomic evaluation.Genet Sel Evol. 2011 May 17;43(1):18. doi: 10.1186/1297-9686-43-18. Genet Sel Evol. 2011. PMID: 21575265 Free PMC article.
-
Genomic prediction in animals and plants: simulation of data, validation, reporting, and benchmarking.Genetics. 2013 Feb;193(2):347-65. doi: 10.1534/genetics.112.147983. Epub 2012 Dec 5. Genetics. 2013. PMID: 23222650 Free PMC article. Review.
Cited by
-
Genomic Analysis, Progress and Future Perspectives in Dairy Cattle Selection: A Review.Animals (Basel). 2021 Feb 25;11(3):599. doi: 10.3390/ani11030599. Animals (Basel). 2021. PMID: 33668747 Free PMC article. Review.
-
Accounting for genetic architecture improves sequence based genomic prediction for a Drosophila fitness trait.PLoS One. 2015 May 7;10(5):e0126880. doi: 10.1371/journal.pone.0126880. eCollection 2015. PLoS One. 2015. PMID: 25950439 Free PMC article.
-
Bayesian genomic models boost prediction accuracy for survival to Streptococcus agalactiae infection in Nile tilapia (Oreochromus nilioticus).Genet Sel Evol. 2021 Apr 21;53(1):37. doi: 10.1186/s12711-021-00629-y. Genet Sel Evol. 2021. PMID: 33882834 Free PMC article.
-
Single-Step Genomic Evaluations from Theory to Practice: Using SNP Chips and Sequence Data in BLUPF90.Genes (Basel). 2020 Jul 14;11(7):790. doi: 10.3390/genes11070790. Genes (Basel). 2020. PMID: 32674271 Free PMC article. Review.
-
Optimized grouping to increase accuracy of prediction of breeding values based on group records in genomic selection breeding programs.Genet Sel Evol. 2019 Nov 15;51(1):64. doi: 10.1186/s12711-019-0509-z. Genet Sel Evol. 2019. PMID: 31730478 Free PMC article.
References
-
- Daetwyler, H. D., B. Villanueva, P. Bijma and J. A. Woolliams, 2007. Inbreeding in genome-wide selection. J. Anim. Breed. Genet. 124 369–376. - PubMed
-
- Dekkers, J. C. M., 2007. Prediction of response from marker-assisted and genomic selection using selection index theory. J. Anim. Breed. Genet. 124 331–341. - PubMed
-
- Falconer, D. S., and T. F. C. Mackay, 1996. Introduction to Quantitative Genetics. Longman, Harlow, UK.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
