

Microsecond simulations of the folding/unfolding thermodynamics of the Trp-cage miniprotein
- PMID: 20408169
- PMCID: PMC3534748
- DOI: 10.1002/prot.22702
Microsecond simulations of the folding/unfolding thermodynamics of the Trp-cage miniprotein
Abstract
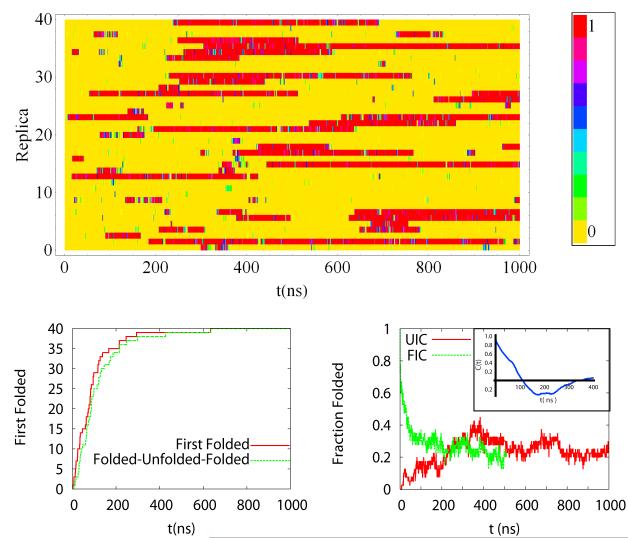
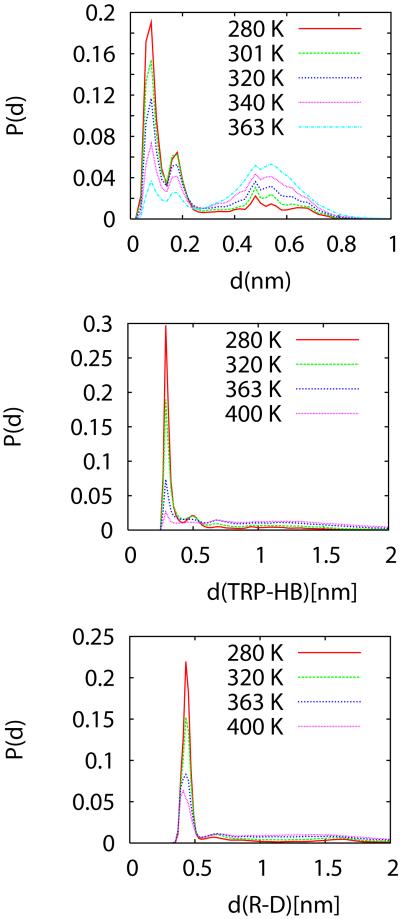
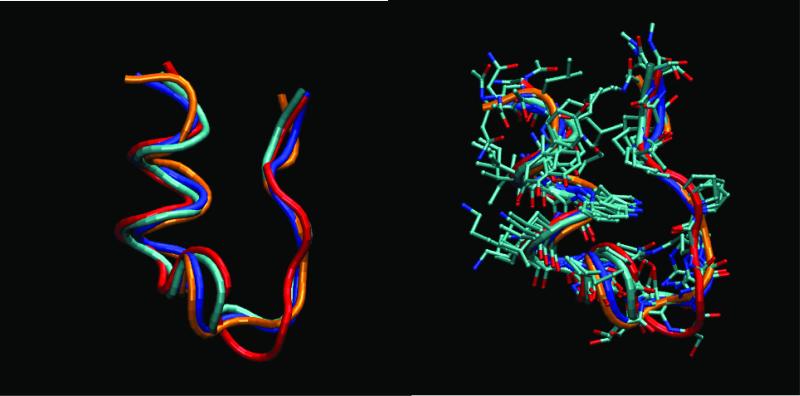
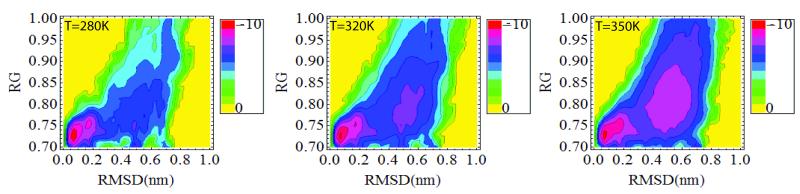
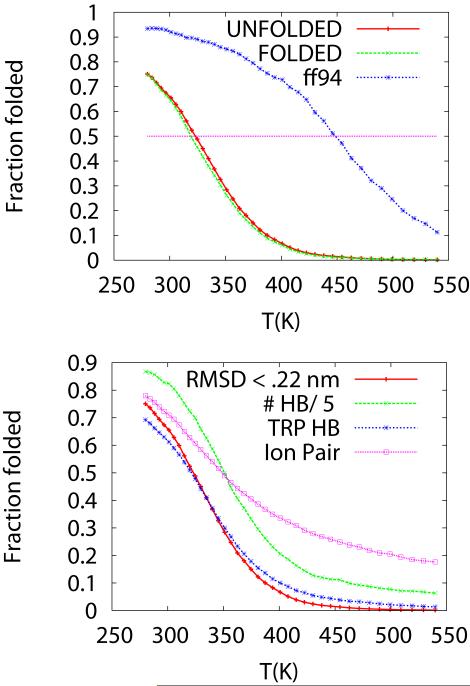
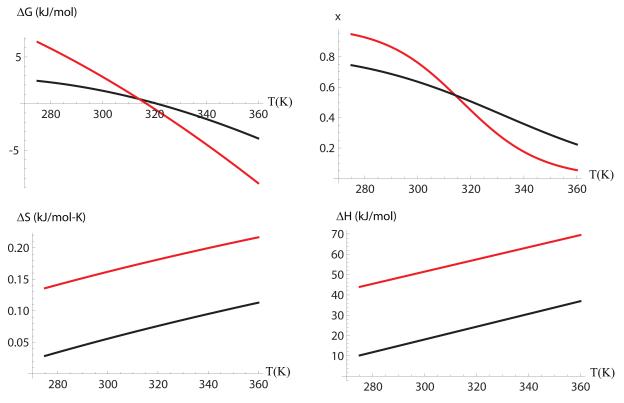
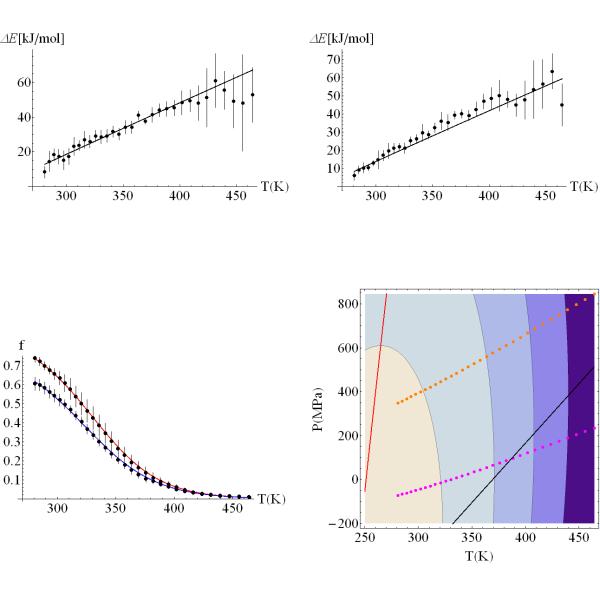
We study the unbiased folding/unfolding thermodynamics of the Trp-cage miniprotein using detailed molecular dynamics simulations of an all-atom model of the protein in explicit solvent using the Amberff99SB force field. Replica-exchange molecular dynamics simulations are used to sample the protein ensembles over a broad range of temperatures covering the folded and unfolded states at two densities. The obtained ensembles are shown to reach equilibrium in the 1 mus/replica timescale. The total simulation time used in the calculations exceeds 100 mus. Ensemble averages of the fraction folded, pressure, and energy differences between the folded and unfolded states as a function of temperature are used to model the free energy of the folding transition, DeltaG(P, T), over the whole region of temperatures and pressures sampled in the simulations. The DeltaG(P, T) diagram describes an ellipse over the range of temperatures and pressures sampled, predicting that the system can undergo pressure-induced unfolding and cold denaturation at low temperatures and high pressures, and unfolding at low pressures and high temperatures. The calculated free energy function exhibits remarkably good agreement with the experimental folding transition temperature (T(f) = 321 K), free energy, and specific heat changes. However, changes in enthalpy and entropy are significantly different than the experimental values. We speculate that these differences may be due to the simplicity of the semiempirical force field used in the simulations and that more elaborate force fields may be required to describe appropriately the thermodynamics of proteins.
Figures
Similar articles
-
Computing the stability diagram of the Trp-cage miniprotein.Proc Natl Acad Sci U S A. 2008 Nov 18;105(46):17754-9. doi: 10.1073/pnas.0804775105. Epub 2008 Nov 12. Proc Natl Acad Sci U S A. 2008. PMID: 19004791 Free PMC article.
-
Protonation/deprotonation effects on the stability of the Trp-cage miniprotein.Phys Chem Chem Phys. 2011 Oct 14;13(38):17056-63. doi: 10.1039/c1cp21193e. Epub 2011 Jul 20. Phys Chem Chem Phys. 2011. PMID: 21773639
-
Folding and unfolding thermodynamics of the TC10b Trp-cage miniprotein.Phys Chem Chem Phys. 2014 Feb 21;16(7):2748-57. doi: 10.1039/c3cp54339k. Epub 2014 Jan 3. Phys Chem Chem Phys. 2014. PMID: 24448113
-
Determination of thermodynamics and kinetics of RNA reactions by force.Q Rev Biophys. 2006 Nov;39(4):325-60. doi: 10.1017/S0033583506004446. Epub 2006 Oct 16. Q Rev Biophys. 2006. PMID: 17040613 Free PMC article. Review.
-
Using simulations to provide the framework for experimental protein folding studies.Arch Biochem Biophys. 2013 Mar;531(1-2):128-35. doi: 10.1016/j.abb.2012.12.015. Epub 2012 Dec 22. Arch Biochem Biophys. 2013. PMID: 23266569 Free PMC article. Review.
Cited by
-
Inclusion of many-body effects in the additive CHARMM protein CMAP potential results in enhanced cooperativity of α-helix and β-hairpin formation.Biophys J. 2012 Sep 5;103(5):1045-51. doi: 10.1016/j.bpj.2012.07.042. Biophys J. 2012. PMID: 23009854 Free PMC article.
-
Sampling of the conformational landscape of small proteins with Monte Carlo methods.Sci Rep. 2020 Oct 23;10(1):18211. doi: 10.1038/s41598-020-75239-7. Sci Rep. 2020. PMID: 33097750 Free PMC article.
-
Atomic-level characterization of the ensemble of the Aβ(1-42) monomer in water using unbiased molecular dynamics simulations and spectral algorithms.J Mol Biol. 2011 Jan 14;405(2):570-83. doi: 10.1016/j.jmb.2010.10.015. Epub 2010 Nov 5. J Mol Biol. 2011. PMID: 21056574 Free PMC article.
-
Forcefield_PTM: Ab Initio Charge and AMBER Forcefield Parameters for Frequently Occurring Post-Translational Modifications.J Chem Theory Comput. 2013 Dec 10;9(12):5653-5674. doi: 10.1021/ct400556v. J Chem Theory Comput. 2013. PMID: 24489522 Free PMC article.
-
A hydrodynamic view of the first-passage folding of Trp-cage miniprotein.Eur Biophys J. 2016 Apr;45(3):229-43. doi: 10.1007/s00249-015-1089-7. Epub 2015 Nov 12. Eur Biophys J. 2016. PMID: 26559408
References
-
- Neidigh JW, Fesinmeyer RM, Andersen NH. Designing a 20-residue protein. Nature Structural Biology. 2002;9(6):425–430. - PubMed
-
- Qiu LL, Pabit SA, Roitberg AE, Hagen SJ. Smaller and faster: The 20-residue Trp-cage protein folds in 4 mu s. Journal of the American Chemical Society. 2002;124(44):12952–12953. - PubMed
-
- Simmerling C, Strockbine B, Roitberg AE. All-atom structure prediction and folding simulations of a stable protein. Journal of the American Chemical Society. 2002;124(38):11258–11259. - PubMed
-
- Zhan LX, Chen JZY, Liu WK. Computational study of the Trp-cage miniprotein based on the ECEPP/3 force field. Proteins-Structure Function and Bioinformatics. 2007;66(2):436–443. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
